Computational Protein Stabilization
Levitate automates a set of Rosetta protein design software protocols as an easty-to-use, SaaS offering proven to reliably identify stabilizing mutations in a wide variety of proteins:
- Correctly predict stabilizing single point mutants
- Category-leading performance in quantitatively calculating energy change upon point mutation
- Experimentally proven protein stabilization algorithms
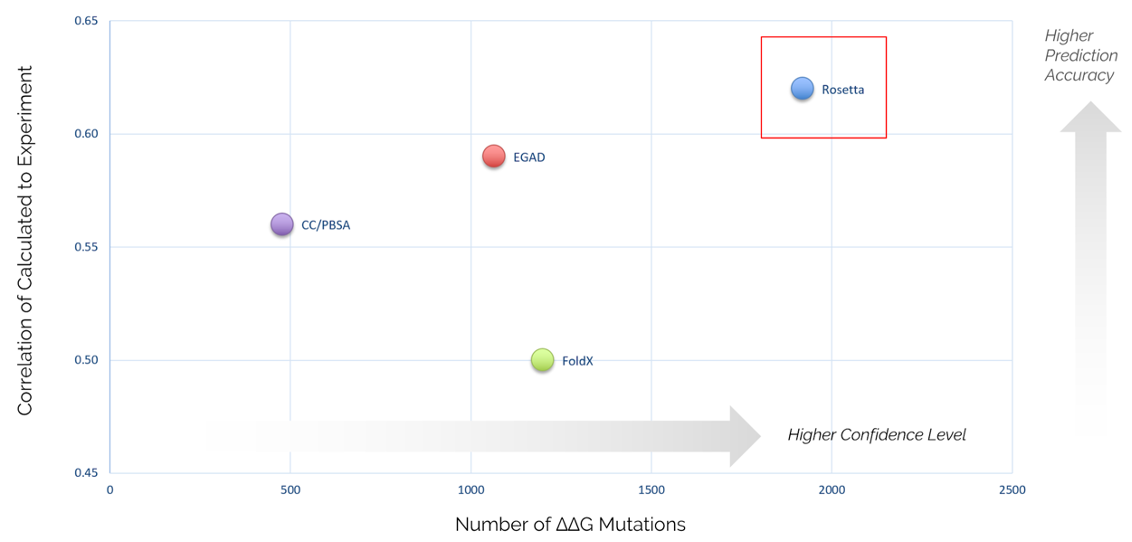
Heighest prediction accuracy and confidence
Only Cyrus delivers software that is refined and tested on large experimental data sets. Our protein mutational free energy predictor has been tested and developed for several years, outperforming similar tools 1.
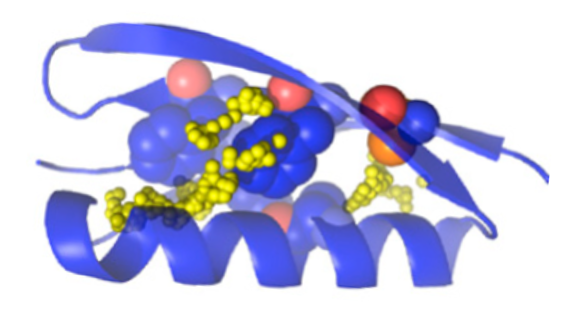
Experimentally demonstrated thermo-stabilization
Thermo-stabilization is often achieved by re-designing a protein core. Our custom-built stabilization protocol has been experimentally validated on a number of proteins 2.
Category-leading performance
Proven success in predicting free energy change upon mutation for a set of roughly 2000 point mutants, with better accuracy and confidence than any other software 1
Experimentally demonstrated thermo-stabilization
- Algorithm to predict stabilizing protein core mutations 2
- Aggressive in silico core re-sampling discovers mutations verified experimentally to stabilize proteins.