Rosetta Structure Sampling
ROSETTA SAMPLING METHODS
Rosetta uses its energy function to determine whether a conformation is favorable. Using this function, it can sample a wide range of conformations in order to optimize protein structure in terms of score. Rosetta uses several ways of sampling conformations. The three primary methods used in our software are: the Monte Carlo (MC) Optimization with the Metropolis Criteria, Gradient-Based Minimization, and Side Chain Rotamer Library sampling. The goal of all sampling methods in to find the conformations with the lowest score.
Traditionally the lowest energy conformation is the one most likely to be the native fold. However, it is only a computational approximation. A true-native will depend on many factors. Proteins evolve under the selective pressure generated by protein function. Most proteins can achieve full function without optimal stability, so our energy functions will never achieve 100% structure prediction. Our energy function can only guide towards a more stable structure.
For example, an enzyme needs to maintain the conformation of its active site, even if that is achieved by allowing some instability. In some cases, protein instability is part of its normal function, such as with Intrinsically Unstructured Proteins. And in many cases, stability is lost if binding partners are not present such as cofactors or dimerization. Therefore optimization in terms of energy will only work if you fully understand a protein’s function and binding partners. Modeling a structure without these partners will find the minimum for the unbound state. A bound state is often very different.
MONTE CARLO
One of the most effective sampling method used in Rosetta is the Monte Carlo (MC) Optimization with the Metropolis Criteria. It is the most common protocol used in Rosetta because it is an effective and flexible way to rapidly sample conformations to find energy minimas.
MC is run by taking a starting structure and iteratively sampling minor changes in structure until it finds a structure with the lowest score. MC makes a random move at every step rather than making an exhaustive sampling of all conformations. This allows it to jump around in conformational space, which is dramatically faster at finding the lowest energy conformation.
Why not do exhaustive sampling?
It is computationally impossible to do an exhaustive search of all possible conformations because of the Levinthal paradox. In 1969, Cyrus Levinthal published a paper showing that a 100 residue protein will have 99 peptide bonds, which would have 198 different Phi-Psi bond angles. If each bond only had three stable angles the number of bond angle variations would be 3,198. Levinthal concluded that it would take longer than the age of the universe for a protein to sample every variation.
Clearly, proteins are able to find their native fold far more rapidly, and from a computational perspective, a shortcut is necessary to find the correct fold.

Cyrus Biotech is named after Dr. Cyrus Levinthal as our software allows you to bypass this paradox to quickly find a correct protein fold.
With each iteration of a MC run a change in conformation, and/or a mutation, is made. Then the energy of the new structure is calculated. The change in conformation is only accepted if the energy of the structure improves. If the energy gets worse there is a probability that the new structure will still be accepted. This potential for the acceptance of unfavorable structures is due to the Metropolis Criteria, which is a probability calculation. The probability (P) tells us whether the change made in this round of MC will be accepted.
P = e–ΔE/kT
Where E = Score (REUs), k = Boltzman constant, T = Temperature
This equation will make it more probable that a change is accepted when the change in energy is small. If the energy of the structure improves then P = 1 so the change is accepted. However if P < 1, there is still a chance of the structure being accepted.
Each round a random number between 0 and 1 is determined. If P < 1 is higher than that number, then the changes made in that round of MC will be accepted.
The example shown below displays one round of MC sampling. In the first stage, there is a local perturbation of the structure. In the second stage the side chains are sampled to find the best conformation given the change. Then the third stage will allow the whole structure to shift based on Minimization of the score function (discussed next). If the change in energy after Minimization is worse than before the perturbation, the change will probably be ignored. This change to the structure will only be accepted if the P < 1 value is greater than the random number, between 0 and 1, that was selected for that round.
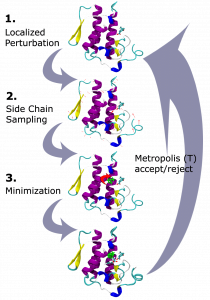
The occasional acceptance of unfavorable energy is helpful for modeling because there are often energetic barriers when moving from one state to another. Putting the structure through unfavorable motions allows sampling outside of the local minima in order to find the global minimum. This allows MC to jump around the energy landscape faster than an exhaustive search, while still primarily making favorable changes.
For more information:
Metropolis N, Rosenbluth AW, Rosenbluth MN, Teller AH, Teller E (1953) Equation of State Calculations by Fast Computing Machines. The Journal of Chemical Physics 21: 1087–1092.
MINIMIZATION
Minimization in Rosetta is a way to find the local energy minima, as shown in the figure below in part A. Instead of using random sampling like Monte Carlo, this tool uses a calculus-based method to only accept changes to structures with scores that are closer to the local energy minimum. This tool only finds the local energy minimum because it does not jump over energy barriers; it can only go down the energy well. This is a gradient-descent minimization of Rosetta energy.
Minimize is a deterministic method so it will always find the same structure. This is different than MC methods which can take many possible trajectories to find different answers with every repeat.
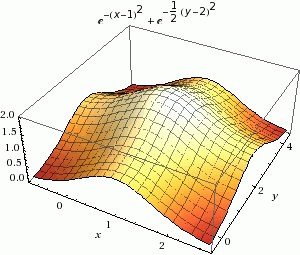
The terms that make up the Rosetta energy function are written in a way that allows us to calculate their first derivative in terms of spatial degrees of freedom. You may remember from calculus that the first derivative is the slope of a curve, which in this case will tell you how to change the conformation of your protein in order to decrease the energy. Though, we are making tiny conformational steps downhill iteratively until it can no longer find a slope downward.
In mathematical terms this is called a Quasi-Newton line search with the Armijo–Goldstein condition using partial derivatives of the Rosetta energy function. This is a variant of the Broyden-Fletcher-Goldfarb-Shanno (BFGS) gradient-based minimization.
ROTAMER SAMPLING
One method for sampling protein conformations is to randomly select a rigid-body side-chain rotamer in a Monte Carlo run. A rotamer is a conformation of a side chain that is commonly found in the PDB and are found to be energetically favorable. Researchers have statistically calculated the propensity of each rotamer in the PDB in order to score more common rotamers better than less common ones. Common rotamers are used to create a library of side-chain conformations.
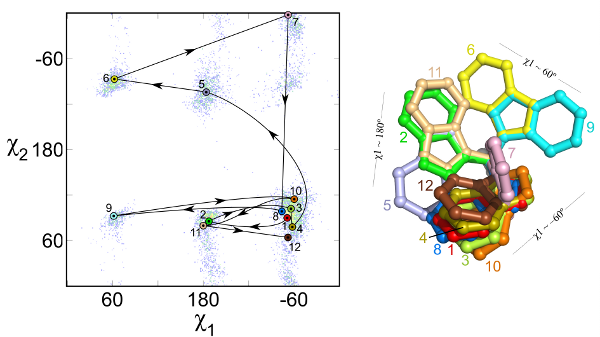
Subramaniam S and Senes A. “An Energy-Based conformer library for side chain optimization: improved prediction and adjustable sampling” Proteins 2012 in press DOI: 10.1002/prot.24111
The rotamer library lists each rotamer’s PDB frequency, its mean torsion angles, and standard deviations as a function of the backbone dihedral angles. It sets this on a bell curve in order to make minimization calculations possible, as these calculations require a continuous function in order to get the first derivative. For each backbone orientation, it is possible to calculate how probable each rotamer is.
Rotamer sampling is different but similar to what happens during rotamer scoring. Many Rosetta protocols allow side-chain conformational changes that randomly shift side chains without rotamers in mind. Then, during the scoring phase, side chains are scored based on how similar a conformation is to a known rotamer.
For more information:
Shapovalov, M.S., and Dunbrack, R.L., Jr. (2011). A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions. Structure, 19, 844-858.
COMBINING SAMPLING METHODS
The power of Monte Carlo optimization is that each iteration can include different methods of sampling. The first step of each iteration usually starts with a random perturbation.
For example, the Relax protocol starts each round with rotamer sampling. Then it runs minimization. This is done with variation of the repulsive energy score term. Ignoring some repulsion allows the structure to make steric clashes that can help the structure cross an energy barrier. Then the repulsion returns to normal to find the new minima in proximity of the new conformation.
Another similar method is called Backrub. In each round of Monte Carlo optimization, it has a perturbation that involves a rotation at a backbone region to rotate the side chain in relation to backbone. Protocols that allow protein mutation often iterate through random mutations and rotamer sampling of the wild type.
SAMPLING METHODS USED IN BENCH
- Prepare: MC Relax
- Repack: MC Side Chain optimization
- Minimize: Minimization
- Relax: MC Relax
- Loop Rebuild: MC with Loop Closure
- Design: MC Side Chain optimization
- Flex Design: MC Backrub
- Relax Design: MC Relax