Advancing Drug Discovery with Rosetta Pocket Protocols and Boltz-2: Finding and Modeling Druggable Binding Sites
In the evolving field of computational drug design, two of the most important challenges are identifying druggable surface pockets on proteins and predicting how strongly small molecules will bind to them. These problems are deeply connected: without knowing where binding is most likely to occur, even the most sophisticated biomolecular complex prediction model may target the wrong site and inaccurately predict compound binding affinity. Conversely, recognizing a promising pocket is often only the beginning when modeling a viable ligand interaction. Levitate Bio offers two powerful complementary tools—Rosetta Pocket Protocols and Boltz-2—to address these challenges. Used in combination, they offer a scalable workflow for drug design that you can leverage to optimize your virtual drug screening.
Discovering Hidden Binding Sites with Rosetta Pocket Protocols
The Rosetta Pocket Protocols are designed to help researchers discover and evaluate potential small-molecule binding sites on protein surfaces. Unlike traditional docking methods that often rely on static structures or guesswork, this approach is rooted in physics-based modeling and ensemble generation.
To use these protocols, we have automated two Levitate Engine APIs: -pocket-measure -pocket-relax. Pocket-measure begins by mapping the protein with a 3D grid that classifies points as protein, surface, or solvent. A potential pocket is identified when a region of solvent is flanked by surface regions—a surface-solvent-surface event—which defines a cavity.
To assess the likelihood that these cavities are biologically meaningful, use the pocket-relax API. Similar to the relax API, this tool leverages a Rosetta algorithm for sampling protein conformational shifts, each scored and evaluated by the Rosetta energy function based on physics and statistics-based terms for packing, solvation, hydrogen bonding, etc. As a result, new terms in the energy function–as well as their weight–will bias conformation sampling. During pocket relax, ensembles of protein conformations are generated both with and without an energetic bias toward pocket formation, mimicking how a protein might fluctuate in the presence or absence of a small molecule. If these biased simulations reveal deep volume pockets that don’t appear in the unbiased relax ensembles—and they do so without major energetic penalties—it suggests that the site is intrinsically accessible and may accommodate a drug-like compound.
By analyzing the biased and unbiased relax results, it becomes clear whether or not a druggable pocket is sampled.
- If the distribution of Rosetta energies for biased and unbiased relax conformations are similar and there are no significant deep-volume pockets discovered, it signifies that druggable pockets don’t form.
- If the distribution of Rosetta energies for biased and unbiased relax conformations is slightly shifted and there are significant pockets discovered, it signifies that druggable pockets form with little energetic consequence.
- If the distribution of Rosetta energies for biased and unbiased relax conformations is significantly shifted and there are significant pockets discovered, it signifies that druggable pockets form only with high energetic costs.
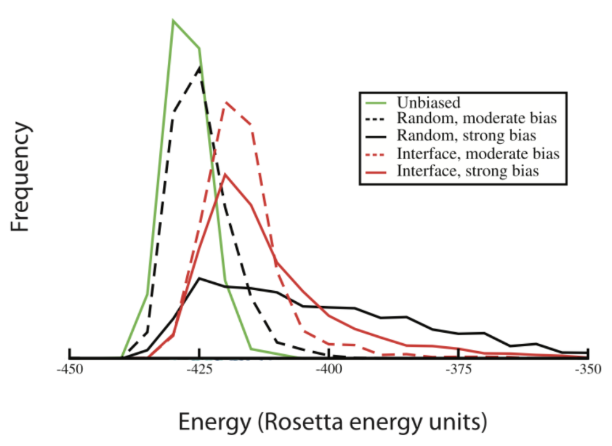
Notably, when energetically favorable pockets form in the absence of a specific ligand, the ensemble of conformations tends to be more diverse, revealing the structural variability that can be exploited in ligand screening.
These outputs—ensemble structures and pocket grids—can serve as the foundation for downstream screening and modeling. Because the method doesn’t rely on a specific compound to define pocket shape, it supports agnostic binding site discovery and facilitates fragment-based or shape-matching approaches.
Modeling Small Molecule Binding and Affinity with Boltz-2
Once a pocket is identified, the next step is to understand how ligands might interact with it—and how strongly. This is where Boltz-2 comes in. Boltz-2 is an ML tool that predicts the structure and binding affinity of protein complexes, including those involving small molecules, nucleic acids, or other proteins. It allows users to steer the modeling process by specifying contact or pocket constraints—such as the residues identified by Rosetta Pocket Protocols—making it possible to focus sampling on the biologically relevant site.
Boltz-2 builds on previous structure prediction models (Boltz-1, Chai-1) and surpasses them in structural accuracy. Additional training and architecture to perform affinity estimation makes Boltz-2 particularly exciting. Boltz-2 is also open-source, unlike AlphaFold3, and can be used commercially, which makes it easier to integrate into drug discovery workflows.
Under the hood, Boltz-2 incorporates data from curated experimental structures, learns from conformational ensembles, and applies constraints grounded in physics to steer the formation of plausible structures. Altogether, Boltz-2 is perfectly positioned to earn a place in established virtual drug screening pipelines alongside more accurate, but considerably slower, approaches like free-energy perturbation (FEP).
However, like all structure-based methods, Boltz-2’s predictions are only as good as its starting assumptions. If the wrong binding site is chosen or if the interface is poorly modeled, the predicted affinity may be misleading. This is why combining Boltz-2 with prior pocket identification using Rosetta can be so powerful. By first identifying a physically accessible and energetically favorable pocket, and then focusing Boltz-2’s modeling on that site, users can substantially improve the plausibility of both the structure and the affinity prediction.
Integrating the Pipeline: From Pocket to Prediction
Use this order of operations to leverage Levitate’s latest drug design APIs:
- Rosetta Pocket Protocols to identify where ligands are most likely to bind based on structural and energetic feasibility.
- Extract those pocket-defining residues and feed them into Boltz-2 as pocket constraints.
- Boltz-2 can then generate plausible protein-ligand complex structures and provide affinity estimates that help prioritize which ligands are most promising.
Together, these tools bring a combination of physical insight and AI-driven modeling to the table. They address two of the most pressing needs in early-stage drug discovery—pocket identification and binding affinity prediction—within a shared framework that can be automated, scaled, and adapted for diverse targets.
Conclusion
The combination of Rosetta Pocket Protocols and Boltz-2 represents a significant step forward in structure-based drug design. By first identifying viable binding pockets through ensemble modeling and then predicting ligand binding strength with an AI-powered engine, researchers can streamline their discovery pipeline with tools that are fast, open, and scientifically rigorous. This approach not only improves the odds of finding a viable lead compound but also ensures that predictions are grounded in structural insight. In drug discovery, as in science broadly, the right starting point makes all the difference—and with these tools, you can start where it counts.
References
David K Johnson, John Karanicolas. Druggable Protein Interaction Sites Are More Predisposed to Surface Pocket Formation than the Rest of the Protein Surface. https://journals.plos.org/ploscompbiol/article?id=10.1371/journal.pcbi.1002951
Saro Passaro, Gabriele Corso, Jeremy Wohlwend, et al. Boltz-2 Towards Accurate and Efficient Binding Affinity Prediction. https://www.biorxiv.org/content/10.1101/2025.06.14.659707v1