AI Folding API
The AI Folding API provides a common entrypoint for a number of AI structure prediction tools. The API currently supports OpenFold, AlphaFold2, ESMFold, and Boltz. Note: This API has been restructured to be better organized and to allow for more tools to be added in the future. The different modes of the API have now been moved to their own subcommands. We have updated the documentation to reflect this change. However documentation for new API commands will be contained within their own pages. Commands previously accessed through the lev engine submit ai-folding command can still be found here.
Command Line Interface
Examples
Model a monomer using AlphaFold2
lev engine submit ai-folding monomer input.fasta \
--ai-tool alphafold
Model multiple monomers in parallel using OpenFold
lev engine submit ai-folding monomer input.fasta input2.fasta \
--ai-tool openfold
Model a monomer using OpenFold with weights trained by DeepMind (AlphaFold2)
Default = OpenFold weights
lev engine submit ai-folding monomer input.fasta \
--ai-tool openfold \
--model-sets alphafold
Create a model using AlphaFold2’s SingleSeq mode with 2 recycles
lev engine submit ai-folding singleseq input.fasta \
--ai-tool alphafold \
--af-n-recycles 2
Create a model using AlphaFold2’s SingleSeq mode with 2 recycles and align all models to the reference structure and report rmsd
lev engine submit ai-folding singleseq *.fasta \
--ai-tool alphafold \
--af-n-recycles 2 \
--reference-structure reference.pdb
Batch create a bunch of models using AlphaFold2’s SingleSeq mode with 2 recycles and align all models to their respective reference structures and report rmsd (Note all files must be in the root directory of the tarballs and the base names must match. I.e. input1.fasta, input2.fasta to input1.pdb, input2.pdb)
lev engine submit ai-folding singleseq fastas.tgz \
--ai-tool alphafold \
--af-n-recycles 2 \
--reference-structure references.tgz
Create a model using AlphaFold Initial Guess
lev engine submit ai-folding initial-guess input.fasta \
--ai-tool alphafold \
--reference-structure initial_structure.pdb
Model a multimer (AlphaFold2 only)
lev engine submit ai-folding multimer input.fasta \
--ai-tool alphafold
Model a monomer using the monomer_ptm weights (AlphaFold2 only)
lev engine submit ai-folding monomer-ptm input.fasta \
Flags
--af-n-recycles(int) (Optional)- Number of recycles to run with AlphaFold2 SingleSeq
--ai-tool(str)- Specify the AI folding tool to run
- Options:
alphafold(default)openfoldesmfold
--exclude-pdb-templates(str) (Optional)- A comma seperated list of pdbids to exclude from being used as templates in alphafold monomer or multimer mode
--existing-model-data(str) (Optional)- Relative path location in GCS of existing model data
--full-db(bool)- Specify to configure
--db_presetflag tofull_dbinstead ofreduced_db(WARNING: may lead to out-of-memory errors) - default =
false
- Specify to configure
--gpu-type(str)- Select the GPU type to use (See Notes for more detail)
- Options:
t4(default)l4a100
--mode(str)- Mode to run with AI tool
- Options:
monomer(default)monomer_ptmmultimersingleseq
--model-sets(str)- The set of model weights to use with OpenFold (See Notes for more detail)
- Options:
alphafoldopenfold(default)
--precomputed-alignments(str) (Optional)- Directory path to precomputed alignments that will be upload and used for AlphaFold2 jobs
--reference-structure(str) (Optional)- Set the initial guess when running AF Initial Guess. Align and report RMSD to this when runing AF SingleSeq.
--run-relax(bool)- Enable or disable the Rosetta relax phase of post-processing
- default =
false
Python Interface
Examples
Model a monomer using AlphaFold2
from engine import EngineClient
client = EngineClient()
client.authorize()
client.submit_ai_folding(
input_files=["input.fasta"],
mode="monomer",
ai_tool="alphafold",
)
Flags
af_n_recycles(int) (Optional)- Number of recycles to run with AlphaFold2 SingleSeq
ai_tool(str)- Specify the AI folding tool to run
- Options:
alphafold(default)openfoldesmfold
exclude_pdb_templates(str) (Optional)- A comma seperated list of pdbids to exclude from being used as templates in alphafold monomer or multimer mode
existing_model_data(str) (Optional)- Relative path location in GCS of existing model data
full_db(bool)- Specify to configure
--db_presetflag tofull_dbinstead ofreduced_db(WARNING: may lead to out-of-memory errors) - default =
false
- Specify to configure
gpu_type(str)- Select the GPU type to use
- Options:
t4(default)l4a100
mode(str)- Mode to run with AI tool
- Options:
monomer(default)monomer_ptmmultimersingleseq
model_sets(str)- The set of model weights to use with OpenFold (See Notes for more detail)
- Options:
alphafold
-
reference_structure(str) (Optional) _ali* Set the initial guess when running AF Initial Guess. Align and report RMSD to this when runing AF SingleSeq. run_relax(bool)- Enable or disable the Rosetta relax phase of post-processing
- default =
false
Outputs
alignments(directory)- Alignment data relevant to AI tool predictions
predictions(directory)- AI tool model predictions
initial_molprobity_reports(directory)- Molprobity report for models output by AI tool
rosetta_relaxed_models(directory)- Rosetta relaxed AI tool models
final_molprobity_reports(directory)- Molprobity report for relaxed models
rmsds- RMSD values for models aligned to a reference structure when run in AF SingleSeq mode
Notes
Model weight sets
alphafold- weights trained by DeepMindopenfold- weights trained by the AlQuarashi Lab for OpenFold
API Post-Processing
The API post-processing protocol consists of the following three steps:
- Generate a molprobity report for the models output from the AI tool
- Idealize and relax the models output from the AI tool with Rosetta
- Generate a molprobity report for the relaxed models.
Modeling large proteins
The amount of GPU memory required increases quadratically with the number of amino acids in the system being modeled. If you are modeling a protein longer than 1500 residues or so, add the following options to the ai-folding submit command: --gpu-type=a100
References
AlphaFold Initial Guess Github
In-depth
Introduction
The field of protein structure prediction was forever changed on the day that Google Deep Mind released the results of their groundbreaking tool, AlphaFold. It was light years better than any other tool, especially for proteins lacking any homolog templates for structure prediction. And two years later, they made another large advancement with AlphaFold2. Many other protein modeling leaders learned from AlphaFold2 algorithms and created their versions. Our AI Folding implements four of these tools, AlphaFold2, AlphaFold-multimer, ESMFold, and OpenFold.
AlphaFold2 and OpenFold have very similar behavior, architecture, and success rates. OpenFold was created by a nonprofit organization funded by donations which trains protein structure prediction tools for purposes that best fit the common good. OpenFold is included here even though it is essentially on par with AlphaFold2 because it is being advanced for other research goals and we expect to implement those additional tools as they become available. ESMFold is provided even though it performs at a lower success rate than AlphaFold2 and OpenFold but it can be 10-fold faster, making it more useful for extremely high throughput needs that don’t require advanced accuracy. AlphaFold-multimer is provided because it is one of the most useful tools available for the prediction of protein-protein interactions.
AlphaFold2, AlphaFold-multimer, AlphaFold Initial Guess and OpenFold
Deep Mind trained AlphaFold2 on protein structures in the PDB so that it can find patterns in how a sequence relates to its 3D structure. It takes a protein sequence as input and tries to predict the physical distance between all amino acid positions in relation to each other. So, given a sequence, it predicts the most probable distance between all positions allowing it to predict their
3D orientation and the bond angles of its backbone. These predictions are iteratively refined and a final structure prediction is made. OpenFold used a very similar architecture as AlphaFold2 and has accuracy on par with AlphaFold2.
AlphaFold-multimer built on AlphaFold2 by retraining it on multimeric protein. Like AlphaFold2, it creates a multisequence alignment (MSA) for the individual proteins, but also looks at how the individual MSAs suggest where a protein-protein interaction exists for the two proteins. So in addition to predicting probability of distances within each protein, it does so between positions on the two proteins. This vastly outperforms tools that do either protein structure prediction of multimers or for tools that look to dock proteins.
AlphaFold Initial Guess is a tool that builds on the AlphaFold2 architecture to create a structure prediction from a starting structure. This is useful for creating a structure prediction for a protein that has been designed to bind a target. The starting structure can be the design model bound to the target and if AF Initial Guess produces a prediction that is close to the design model, it is a good indicator that the design model is correct.
Evolutionary Structure-based MSA-Informed Folding (ESMFold)
ESMFold has a very different architecture than AlphaFold2, AlphaFold-multimer, and OpenFold. It was also trained on the PDB and uses MSAs to understand evolutionary relationships for a protein. But ESMFold calculates residue-residue coupling indicating that positions coevolved. It determines which positions more directly interact by correlating the degree of coevolution. Then it treats interacting residues like protein fragments and samples their interactions. It randomly samples protein fragment orientations by Monte Carlo (a bit like trial and error). Iterative fragment sampling to optimize the residue-residue coupling leads to a final structure prediction.
Confidence scores
For AlphaFold2, AlphaFold-multimer, OpenFold, and ESMFold output models are given a confidence score for every position of the structure prediction. This is a great indicator of how accurate that the prediction is. Like all structure prediction tools, they will always create a prediction. But the score will tell you if its prediction has a good chance of being meaningful. The score for each position of the structure prediction can be found in the PDB file in the B Factor column for the unrelaxed structure. Relaxed structures will not retain that information.
Example: The score for residue 27 (Serine) is 98.61

High pLDDT scores are better. People commonly use 70 to 80 as a cutoff to consider a prediction to have high accuracy. Structure viewers such as pymol can color a structure by B Foctors. For pymol, you can select A (for Action) in the right window to the right on the structure name and choose preset and ba factor putty, as shown below. High confidence will be in red and lowest confidence in blue.
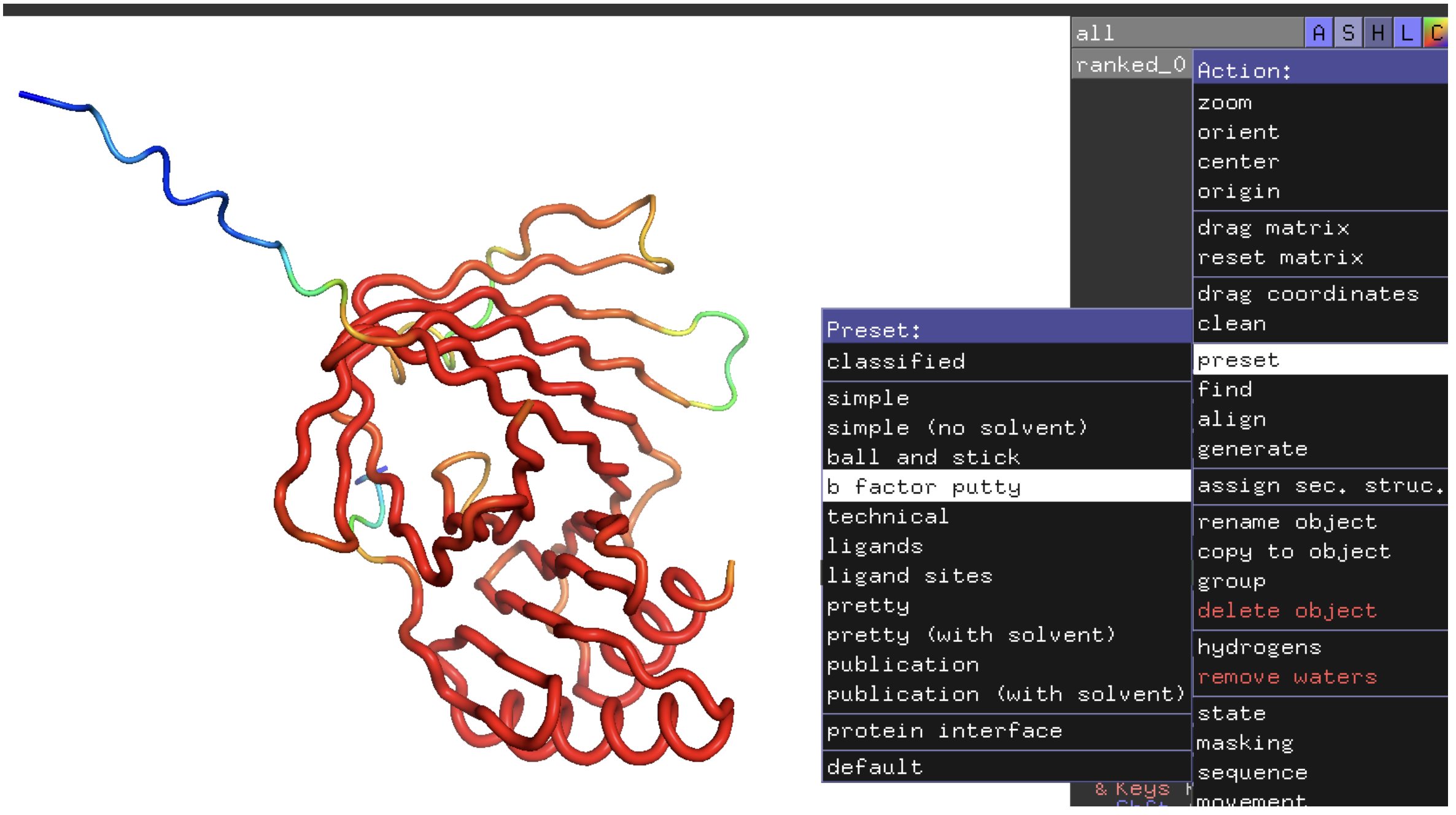
In that example, the blue region is an unstructured, soluble part of the protein. You can often predict which portions of a protein are unstructured before doing a structure prediction and it is best to leave it out of the prediction.
Simple example
AI Folding’s simplest example will take an input sequence of a monomeric protein in fasta format and output a structure prediction in a PDB file.
The command for running a simple protein structure prediction:
lev engine submit ai-folding monomer input.fasta
input.fasta- Any file name that comes after
monomeris expected to have the protein sequence that will be used to generate a new structure. This can be any name followed by .fasta.
- Any file name that comes after
By default, it will run AI Folding in monomer mode and will use AlphaFold2.
Output files
There are 6 possible output files, depending on which flags were used:
inputswill be a directory with the fasta file used as input.JOB_INFO.txtwill have the details of how the job was run including name of the input fasta file, mode (single or multisequence), tool (AlphaFold2, OpenFold, or ESMFold), OpenFold model if relevant, gpu type, wether the prediction was Relaxed, and number of recycles (refinement iterations).outputs(when you run AlphaFold2) orpredictions(when you run AlphaFold2, OpenFold, or ESMFold)** will be a directory with the structure prediction named the same as th input fasta followed by .pdb.rosetta_relaxedwill be a directory with a Relaxed model of your structure prediction.alignments(when you run OpenFold) will be a directory with files showing identities and alignments for an MSA.rmsdswill be a file with RMSD values for models aligned to a reference structure when run in AF SingleSeq mode.
Note that the the relaxed PDB can be opened in a text file if you want to see the Rosetta score for the whole structure and the individual residues broken down into individual score terms. This is very useful for predicting relative stability of a structure when running design with the same input structure.
Running relax after structure prediction
A structure prediction can be Relaxed automatically after the structure prediction is run which is useful both for optimizing the structure and because the relaxed PDB will have Rosetta scoring details in the files listed at the end of the file. The full breakdown of all score terms will be shown for the whole structure and for individual positions.
lev engine submit ai-folding monomer input.fasta --run-relax
Running monomer structure prediction with alternate tool
You can run OpenFold or ESMFold when generating a structure prediction for a monomer by adding a flag as shown below:
lev engine submit ai-folding monomer input.fasta --ai-tool openfold
-
--ai-tool openfold (or esmfold or alphafold)- Flag needed to define AI folding tool OpenFold or ESMFold. You can use this to define AlphaFold2 or it will select it by default.
Running AlphaFold2 or OpenFold without a multi-sequence alignment (MSA)
An MSA is used by default when you run AlphaFold2 and OpenFold because it greatly improves accuracy to use evolutionary information for structure prediction. However, the reverse is true when running a structure prediction for a sequence that was not evolved such as engineered proteins. Running either tool in single sequence mode requires the flag shown below:
lev engine submit ai-folding singleseq input.fasta
-
singleseq- Subcommand needed to run in single sequence mode
Running AlphaFold-multimer
lev engine submit ai-folding multimer input.fasta
-
multimer- Subcommand needed to run in multimer mode
Unlike the other AI Folding tools, there will be 25 structure predictions run because multimer predictions are more challenging. The structures will be ranked by confidence score, pLDDT. a large number of output models generated because protein-protein interactions are far more challenging to accurately predict so multiple variations will be generated and ranked.
Glossary of AI Folding command-line flags
-
--ai-tool openfold (or esmfold or alphafold)- Flag to define AI folding tool OpenFold or ESMFold. You can use this to define AlphaFold2 or it will select it by default.
-
monomer (or multimer, singleseq, or initial-guess)- Subcommand to specify the mode. AlphaFold2 can be run in multimer mode. It can also be run without using the multisequence alignment (singleseq) which is best when doing structure prediction of designed sequences. It can also be run in initial-guess mode which biases the prediction towards a starting structure and can be useful for validating a design model.
-
--run-relax- Flag to run relax on structure optimization. This will find a more favorable conformation for the protein and output Rosetta scoring for the structure at the end of the output file. Open the relaxed pdb file in a text reading application to read the scores.