ProteinMPNN API
The ProteinMPNN API provides an interface to the ProteinMPNN protein design tool. This tool takes as input a PDB file and rapidly generates new sequences predicted to fold to the backbone of the input PDB.
Command Line Interface
Examples
Predict a single new sequence for an input PDB
lev engine submit protein-mpnn 1ubq.pdb
Predict 1000 new sequences for an input PDB
lev engine submit protein-mpnn 1ubq.pdb \
--n-mpnn-designs 1000
Predict 1000 new sequences for an input PDB using an elevated temperature (default temperature is 0.1)
lev engine submit protein-mpnn 1ubq.pdb \
--n-mpnn-designs 1000 \
--sampling-temperature 0.2
Create De Novo sequences for part of a protein structure
lev engine submit protein-mpnn input.pdb \
--n-mpnn-designs 50 \
--fixed-residue-positions="A1-10 A22 A24 A26 A220-250 B30-60"
Score the sequences in a fasta file without generating new sequences
lev engine submit protein-mpnn input.pdb \
--fasta-file input.fasta \
--score-only true
Fix residues regions that are not poly-glycine from RFDiffusion
lev engine submit protein-mpnn input.pdb \
--poly-gly
Upload structures in batch to protein-mpnn
# All PDB files in the current directory using wildcard
lev engine submit protein-mpnn *.pdb
# All PDB files compressed and zipped into a tarball
lev engine submit protein-mpnn input.tgz
Flags
--batch-size(int) (Default:100)- The number of PDB files to process in each batch. This may be useful when you have a large number of PDB files to process
--fasta-file(str) (Optional)- The path to a fasta file containing sequences to score. This should be used in conjunction with
--score-only
- The path to a fasta file containing sequences to score. This should be used in conjunction with
--fixed-residue-positions- The residues which will not be designed, space-separated list of positions in the format {chain}{startres}-{endres}
--generate-af2mm-fasta(bool) (Default:true)- If set to true the model will generate fastas compatible with AlphaFold2 Multimer
--gpu-type(str) (Default:t4)- The GPU to run the model on. Set this to
a100if you are generating a very large number of sequences - Options:
t4a100
- The GPU to run the model on. Set this to
--min-gly-threshold(int) (Default:3)- The minimum number of continuous glycine residues to create fixed residue selections. Default is 3. Active with
--poly-gly
- The minimum number of continuous glycine residues to create fixed residue selections. Default is 3. Active with
--n-mpnn-designs(int) (Default:1)- The number of sequences to design, default is 1
--pdb-file(str) (Required)- The path to a PDB file containing the protein backbone you want to design sequences for
--poly-gly(bool) (Default:false)- If added to protein-mpnn command, this will fix residues regions that are not poly-glycine from RFDiffusion. This can be used in conjuction with
--min-gly-threshold
- If added to protein-mpnn command, this will fix residues regions that are not poly-glycine from RFDiffusion. This can be used in conjuction with
--sampling-temperature(float) (Default:0.1)- The sampling temperature of the model, default is 0.1, higher values will result in more variation of output sequenes
--score-only(bool) (Default:false)- If set to true the model will score the sequences in the fasta file and not generate new sequences
--tied-positions(str) (Default:"")- The list of positions on each chain that should be symmetrically tied together (only if not already set by the symmetric chains flag). Individual sets should be separated by a space. Chains in the set should be separated by a
/i.e'A1-5/B1-5 C5-20/D5-20'
- The list of positions on each chain that should be symmetrically tied together (only if not already set by the symmetric chains flag). Individual sets should be separated by a space. Chains in the set should be separated by a
Python Interface
Examples
Predict a single new sequence for an input PDB
from engine import EngineClient
client = EngineClient()
client.authorize()
job_id = client.submit_protein_mpnn(
pdb_path="input.pdb"
)
Flags
batch_size(int) (Default:100)- The number of sequences to design at a time
fasta_file(str) (Optional)- The path to a fasta file containing sequences to score. This should be used in conjunction with
--score-only
- The path to a fasta file containing sequences to score. This should be used in conjunction with
fixed_residue_positions(str) (Optional)- The residues which will not be designed, space-separated list of positions in the format {chain}{startres}-{endres}
gpu_type(str) (Default:t4)- The GPU to run the model on. Set this to
a100if you are generating a very large number of sequences - Options:
t4a100
- The GPU to run the model on. Set this to
pdb_path(str) (Required)- Input PDB to design sequences for
sampling_temperature(float) (Default:0.1)- The sampling temperature of the model. Higher values will result in more variation of output sequences
score_only(bool) (Default:false)- If set to true the model will score the sequences in the fasta file and not generate new sequences
sequence_per_target(int) (Default:1)- The number of sequences to design per target
Outputs
designed_sequences.fasta- A FASTA file containing all designed sequences. The first record in the file is the native sequence of the protein in the PDB file. The headers of the FASTA file contain score and sequence recovery values for each designed sequence
In-depth
Introduction
ProteinMPNN stands out as a groundbreaking sequence design protocol leveraging deep learning. It can generate sequences by inputting a protein or protein complex file, replacing the entire structure or specific sections. With a remarkable sequence recovery of 52.4%, surpassing classic Rosetta’s 32.9% (fixed backbone version), it demonstrates an enhanced understanding of proteins, surpassing Rosetta’s traditional physics, statistics, and Monte Carlo-based approaches when re-engineering a whole protein structure. Wet lab validation has successfully expressed various protein types, including monomers, oligomers, nanoparticles, active enzymes, and target-binding proteins.
Protein Message Passing Neural Network (ProteinMPNN) is trained to take a structure and predict what sequence is most likely to occur at every position given the structural features. It is a significant improvement over Rosetta when you intend to redesign a large section of the entire sequence of a protein. This has long been a challenging goal because full redesign commonly results in highly insoluble protein expression.
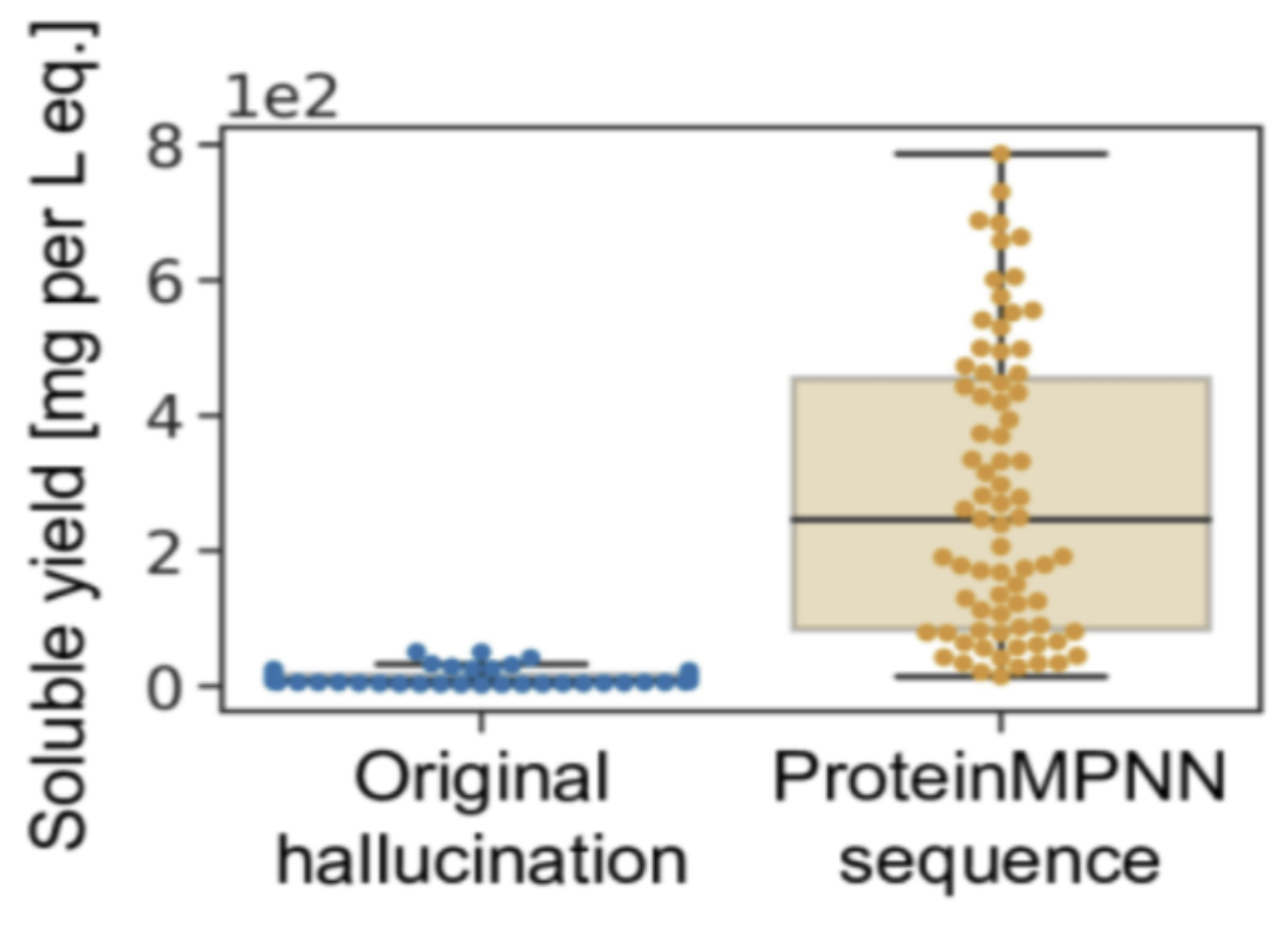
In the initial study, researchers employed ProteinMPNN to re-engineer 150 proteins. Out of these, 73 exhibited soluble expression in E. coli, boasting a median soluble yield of 247 mg per liter. The figure below illustrates the yield, offering a comparative analysis with designs generated by AlphaFold (referred to as Original Hallucination in this context). While it’s important to note that AlphaFold wasn’t specifically designed for protein engineering, making it an imperfect benchmark, the remarkable solubility and yield of the designs remain noteworthy.
Simple example
ProteinMPNN in its simplest form will take an input PDB that contains a protein and will output a fasta file with a completely new sequence for the whole structure that it predicts, if expressed, would fold into a protein with that backbone conformation. It is standard practice to generate many designs because it is capable of generating incredible diversity even for a small peptide. It is common for people to have ProteinMPNN output anywhere from 50 to 50,000 sequences.
lev engine submit protein-mpnn input.pdb --n-mpnn-designs 50
-
input.pdb- Any file name that comes after ‘protein-mpnn’ is expected to have the protein structure that will be used to generate a new sequence. This can be any name followed by .pdb.
-
--n-mpnn-designs 50- Flag needed to define # of output sequences as 50
There will be 3 outputs from this run:
inputswill be a directory with the PDB file used as input which will be named pdb.pdb.JOB_INFO.txtwill have the details of how the job was run including name of the input PDB, number of repeats, sampling temperature, GPU type that ran the job, and positions in the PDB that were fixed (not allowed to change sequence).designed_sequences.fastawill have the output sequences and qualitative information about the run which will help you understand the confidence level of the run. Here is an example:

score- This is a measure of how good the sequence is expected to be for the protein conformation. Lower is better.- It is averaged for all residues and chains.
- The input sequence and the designed sequences are scored to help indicate how much better the designs are predicted to be.
- In machine learning terms, it is a negative log probability of sampled amino acids and can be thought of as a loss function.
global_score- This is the same as the score, but is an average of all chains.fixed_chains- chains that were not designed (fixed)designed_chains- chains that were redesignedmodel_name- ProteinMPNN model version that was used to generate resultsgit_hash- github version that was used to generate outputsseed- This is a random number that is generated every run to create randomness for designT=0.1- T is the temperature. Increasing it will create more diversity in the sequence output but is associated with less sequence recovery (likelihood of generating native sequences).sample- Output designs are numbered (1, 2, 3…etc).seq_recovery- percent identity to input
Create De Novo sequences with more diversity
By default, ProteinMPNN will run at a low temperature which mean that it will predict sequences that are closer to what it thinks native sequence should be. You can allow it to diverge more by increasing the temperature from its default 0.1. Common values range from 0.2 to 0.5, but can be done up to 1. In this example, we repeat the last command, but change the temperature to 0.25:
lev engine submit protein-mpnn input.pdb --n-mpnn-designs 50 --sampling-temperature 0.25
Create De Novo sequences for part of a protein structure
ProteinMPNN can be run for one or more sections of an input structure. This is done by listing the positions that you wish to retain the sequence found in the input PDB. The syntax for specifying those residues is {chain}{startres}-{endres} for each block to keep fixed with a space between noncontinuous positions. Here is an example for a redesign of a PDB with chains A and B where everything is designed except positions chain A1-10, 22, 24, 26, 220-250 and in chain B30-60.
lev engine submit protein-mpnn input.pdb --n-mpnn-designs 50 --fixed-residue-positions="A1-10 A22 A24 A26 A220-250 B30-60"
A method for fixing positions without manually specifying them is --poly-gly mode, where --min-gly-threshold sets a minimum number of continuous glycine residues that can appear in the sequence to qualify as a designable selection. From these designable selections, nondesignable or fixed positions will be created. This is useful for one input PDB or a batch input of multiple PDBs or a tarball, typically the outputs of RFDiffusion. For batch mode, the wildcard * in *.pdb will select all files that end in .pdb from the current directory. You can also pass input.tgz containing PDB files for batch mode.
lev engine submit protein-mpnn *.pdb --n-mpnn-designs 10 --poly-gly --min-gly-threshold 3
Create a tarball of PDB files from an RFDiffusion output
First, move into the directory containing the RFDiffusion outputs using cd (change directory command). This is important to ensure that you don’t overwrite files with duplicate names. Then, use the tar archiving command to compress and zip the PDB files. The flags -czvf will create (c), name (f), zip (z), and enable verbose output (v) of your tarball.
cd rf-diffusion-jobID/out
tar -czvf protein-mpnn-input.tgz *.pdb