Antibody HM API
OVERVIEW
HM Antibody is a structure prediction tool for the variable region of antibodies. The protocol assumes that your sequence is a naturally-occuring antibody, which means that there is a limit to how much your input can diverge from the norm. Often, users have antibody designs that significantly lengthen a CDR loop. This is not possible with HM Antibody. In that case, use regular HM. HM will need you to run the Heavy and Light chains separately.
BACKGROUND
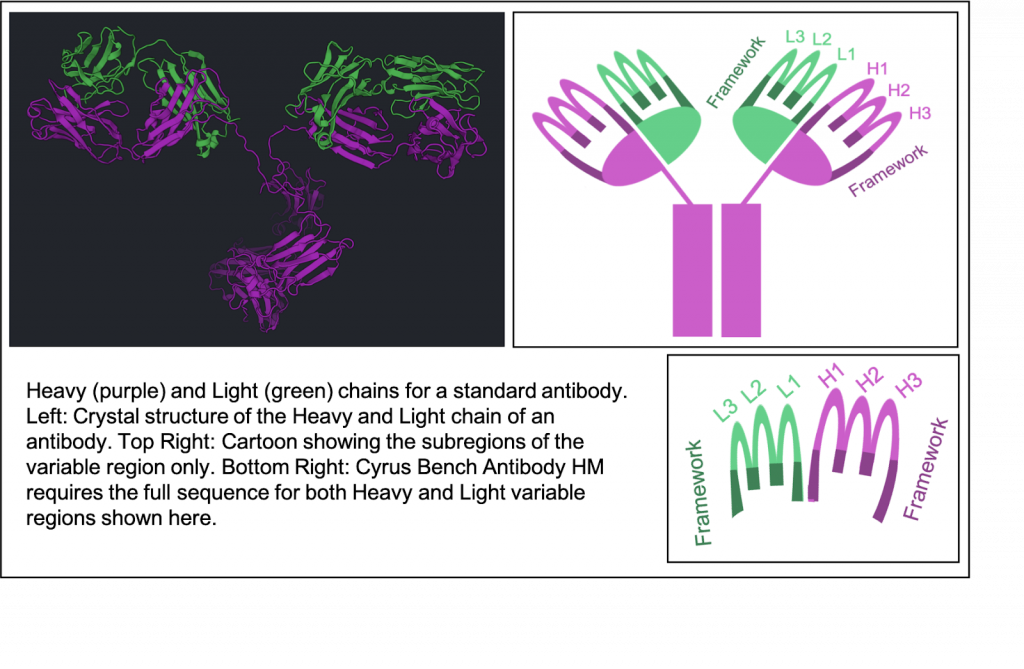
Antibody Homology Modeling uses the RosettaAntibody homology modeling tools developed by Jeffrey Gray’s lab at Johns Hopkins University to generate structural homology models of Fv domains of antibody [1]. This algorithm first identifies the antibody standard components, which include framework regions, light-to-heavy (LH) chain orientations, and heavy or light chain CDR loops [2] from the input sequence. For each of these antibody components, the best template structures are identified from the PDB by sequence homology and are modelled independently. Each of the frameworks and loops are then put together to finally generate low energy homology models of the complete Fv domain of the antibody structure.
HOW TO RUN HM ANTIBODY
Input: Sequence Submission
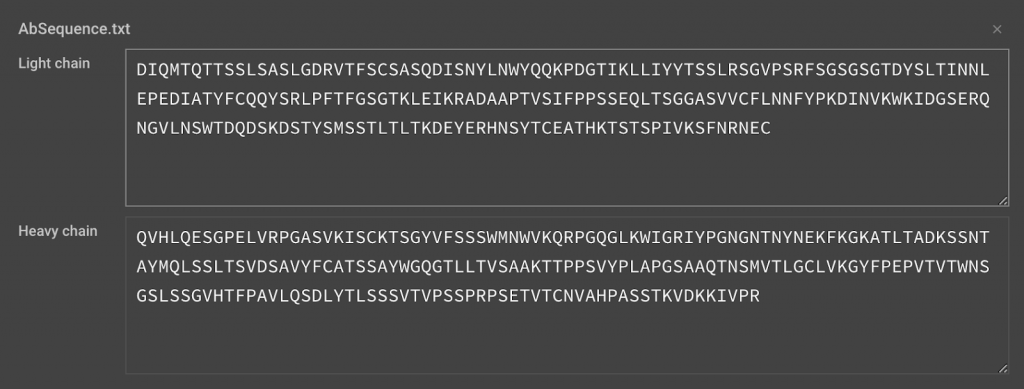
Your input sequence should be the variable regions of the Heavy and Light chains only.
Click the button called “Antibody HM” located in the right hand panel under “Structure Prediction” in Bench. Two input sequences are required for antibody modeling, which includes the light and heavy chain sequences. Note that only the Fv domain portions of the full length antibody sequence for heavy and light chains will be modelled. Once, both fields are filled in with the sequences, click on the green “Run” button to submit the job for generating the antibody models.
![]()
Alternatively, you can upload your sequences in a fasta file by clicking . We are currently limited to one pair of input sequences at a time. Your input file should list the Heavy Chain first followed by the Light Chain. Any further lines will not be included.
Example:
>Heavy Chain
- QVHLQESGPELVRPGASVKISCKTSGYVFSSSWMNWVKQRPGQGLKWIGRIYPGNGN TNYNEKFKGKATLTADKSSNTAYMQLSSLTSVDSAVYFCATSSAYWGQGTLLTVSAA KTTPPSVYPLAPGSAAQTNSMVTLGCLVKGYFPEPVTVTWNSGSLSSGVHTFPAVLQ SDLYTLSSSVTVPSSPRPSETVTCNVAHPASSTKVDKKIVPR
>Light Chain
- DIQMTQTTSSLSASLGDRVTFSCSASQDISNYLNWYQQKPDGTIKLLIYYTSSLRSG VPSRFSGSGSGTDYSLTINNLEPEDIATYFCQQYSRLPFTFGSGTKLEIKRADAAPT VSIFPPSSEQLTSGGASVVCFLNNFYPKDINVKWKIDGSERQNGVLNSWTDQDSKDS TYSMSSTLTLTKDEYERHNSYTCEATHKTSTSPIVKSFNRNEC
Verify that the Heavy and Light chains are not switched before clicking Run.
Output: Structure Visualization and Analysis
Each antibody model takes approximately 3-4 minutes to generate a rough model, and an additional 16-18 minutes to produce a subsequent relaxed high quality model. A typical antibody (heavy and light chains that are around 220 amino acids in sequence length) run will take approximately 3-4 hours and will produce ten structures low energy structures (relaxed) of the Fv domain (heavy and light chain) of your input antibody sequence.
Additional modeling will often help optimize an H3 loop. This is usually longer than other loops and more diverse. We recommend running Loop Rebuild on the H3 loop. Instructions can be found here.
METHOD OVERVIEW
Step One: Search for Homologs to use as Structural Templates
We use your Light and Heavy chain sequences to detect the CDRs (L1, L2, L3, H1, H2, H3), the Framework (FRL, FRH), and predict the orientation between the heavy and light chain. Each of these 8 subregions are individually identified by comparing them to known subregions that can be used as homologous templates for the corresponding subregion. The orientation between heavy and light chains is also predicted by comparing to known antibodies.
Homolog templates are not just discovered by sequence alignment. A profile has been created for all potential templates so that a more informative comparison can be made. Potential template profiles are HMMs (Hidden Markov Models) which were created using the ANARCI (Antigen receptor numbering and receptor classification) tool from a large database of antibodies. The database is divided into subregion in order find the matching component in your protein. We also use ANARCI to create an HMM of your sequence. Your HMM is compared to all HMMs to find the strongest regions of similarity. Then your sequence will be aligned with its corresponding templates.
If this protocol is unable to identify one or more subregions, you will get an error message indicating that you are missing a necessary component. It is possible that your sequence just varies too far from the norm for the protocol to identify all subregions. Do not include sequences outside of this variable region. This is what our protocol expects:
Framework1 – CDR1 – Framework2 – CDR2 – Framework3 – CDR3 – Framework4
That is the sequence order found in naturally-occurring antibodies.
Step Two: Thread your Sequence on Structural Templates and Paste together
Each region was identified and compared to a database of structures of that region. The best hit for each is used to thread the sequence over the structure in the predicted region. Since the subregions are threaded into different template, these subregions are pasted together before further modeling. The orientation between the Heavy and Light is determined by sequence similarity of both chains to a database of Heavy/Light chains with known orientation.
Step Three: Modeling Fine Details Once your sequence is threaded over the templates and stitched together, the orientation between heavy and light chain can move the protein into its initial modeling conformation. Then Rosetta structure optimization can take place. This takes into account the physics of the sequence variation that differs from the templates in order to find an energetically favorable orientation. While the structure will be biased by the templates, this will allow the fine details to find a better shape. This protocol has a high success rate, but does not take into account the effect of a binding partner. Antibodies often have significant conformational changes when binding their antigen. So further modeling in Bench in the presence of the antigen may find the true antibody pose.
METHOD ASSESSMENT
Methods for modeling antibodies have been tested with the Antibody Modeling Assessment (AMA). This is a structure prediction competition which provides sequences of antibody variable regions, but holds the known structures secret until modeling methods can provide their predictions. Rosetta’s Antibody tool was tested along with CCG, PIGS, and Accelrys in the first AMA (2011). All methods were able to predict backbone structure at around 1.2 Å RMSD. H3 loops are the most challenging and predictions were around 3 Å RMSD. The second AMA (2014) tested 7 modeling methods. The Rosetta method (Gray Lab) was greatly improved since the first version and did much better in AMA II. For non-H3 loops, 42 of the 55 CDRs and all framework regions were predicted under 1 Å RMSD. For the eleven H3 loops, Rosetta had the best prediction for four targets.
Rosetta Antibody is among the top tools for modeling antibody structure and in many cases it is superior. Its predictive power has improved since the last AMA because there are many more homologs found in the PDB and better predictions can be found using non-antibody proteins as structure templates. This is particularly important for H3 regions, which are known to be longer and more variable.
REFERENCES:
-
Weitzner BD, Jeliazkov JR, Lyskov S, Marze N, Kuroda D, Frick R, Adolf-Bryfogle J, Biswas N, Dunbrack RL Jr, Gray JJ. Modeling and docking of antibody structures with Rosetta. Nat Protoc. 2017 Feb;12(2):401-416. doi: 10.1038/nprot.2016.180. Epub 2017 Jan 26
-
Dunbar J, Deane CM. ANARCI: antigen receptor numbering and receptor classification. Bioinformatics. 2016 Jan 15;32(2):298-300. doi: 10.1093/bioinformatics/btv552. Epub 2015 Sep 30.