Relax
Relax is one of the most used Rosetta methods for sampling conformational changes in a protein. It is more aggressive than Minimize, so it tries out orientations that are up to a few angstroms away from the starting structure.
RUNNING RELAX
Select the folder that contains the structure or structures that you want to Relax. This will bring the structures in that folder into the center window. Select all the structures that you want to Relax by clicking in the box to the left of their names. Next, click the ‘Relax’ button on the top right under Actions to initialize Relax in the center window. Select the number of repeats you would like to run and click ‘Save & Run’.
HOW MANY REPEATS OF RELAX SHOULD I DO?
It is recommended to do many repeats of Relax in order to find the most energetically favorable conformation of your protein. Every repeat of Relax has a different trajectory because it uses a different random number to select the trajectory path. In order to determine how many repeats are needed, we recommend starting with 5 to 50 repeats. A small protein (under 50 residues) will usually converge easily with 5 repeats. A larger protein (>1,000 residues) will need 50 repeats. Then the best structure of those 50 can be used as a starting structure for another 50 repeats. This may need to be repeated several times.
Below is an example. This 219 residue protein will easily find its minimum in two rounds of repeats. The first round of 50 repeats reduced the score by 323 or more REUs. Using the structure with the lowest score (that was 337 lower than the original input), a second round of repeats could reduce the score another 3 REUs. Further repeats did not generate improvements.
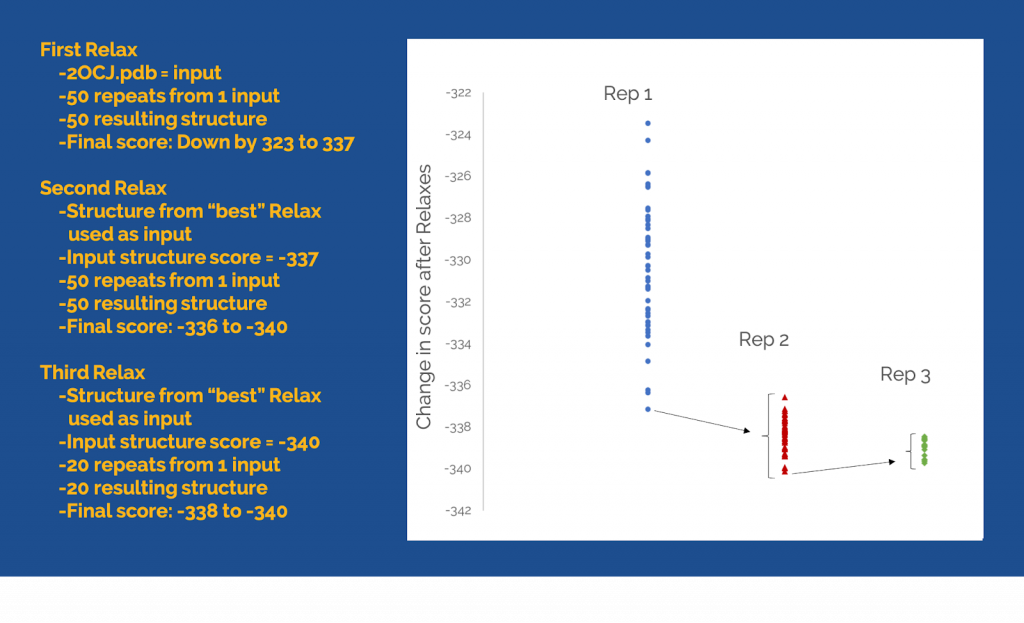
A Relax run will always sample the same number of conformations. Sometimes, this is not enough sampling to find the lowest score, so we need to keep sampling more conformations in order to find the best one. For example, if you run 50 repeats of Relax. You will need to rank the 50 output structures by score. If the lowest energy structure is lower than the starting structure, then you may be able to continue improvement of the conformations. Use the best structure from the Relax as a starting structure for another 50 repeats of Relax. Again, rank results by score and compare to the input structure for this Relax. Repeat this process until the score no longer gets better.
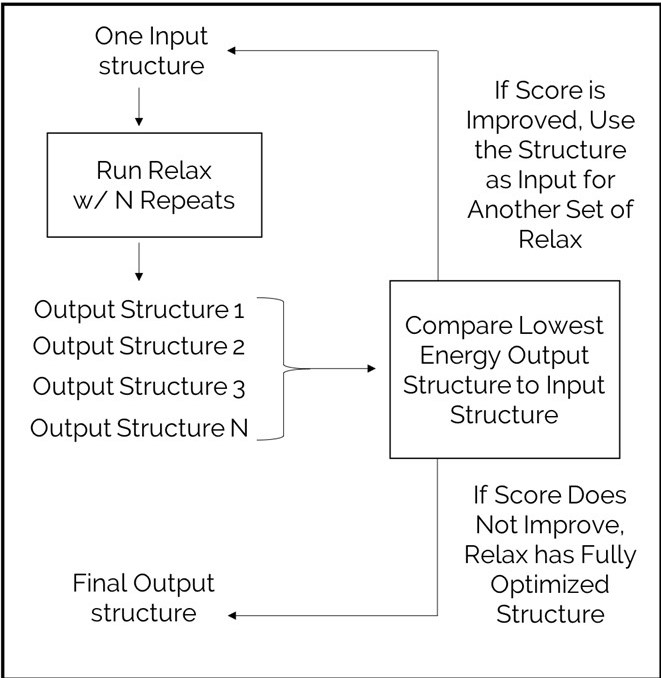
The structure will need more rounds of Relax if the original structure is large, flexible, and/or is far from the optimal conformation. The final structure will only represent a native fold if the original structure is within a few angstroms of native.
WHAT DOES RELAX DO?
This is a Monte Carlo (MC) method of sampling conformational changes in your protein. The MC algorithm makes changes to the protein’s structure, then the energy of the new conformation is calculated in order to compare it to the energy before the change. If the energy is better, then the change is accepted, but if the energy gets worse there is still a chance that the change is excepted. Occasionally allowing moves that are unfavorable allows the structure to get over an energetic hurdle to find a new minima.
Each iteration of the MC cycle goes through a perturbation of the protein’s structure by Repacking, then Minimizing. It measures energy using the full-atom Rosetta energy. One of the things included in Rosetta energy is the repulsive forces between atoms. Repulsion is varied every four rounds so that it starts very low and increases to 100% by the fourth round. Decreased repulsion allows more clashing so that energetic hurdles can be overcome in order to find new minima. Ramping repulsion back to 100% allows it to avoid paths that don’t lead to new minima. So a cycle of Monte Carlo includes four rounds of Repack-Minimize with ramping repulsion followed by acceptance or denial of the new conformation.
REFERENCE
Conway P, Tyka MD, DiMaio F, Konerding DE, Baker D. Relaxation of backbone bond geometry improves protein energy landscape modeling Protein Sci. 2014 Jan;23(1):47-55.