Replace Ligand
OVERVIEW
The Replace Ligand tool allows you to swap a new ligand into a pocket in place of an existing ligand. This is useful when you want to dock a molecule that is similar to a ligand with a known binding conformation. Design and other modeling can then be carried out around the replaced ligand.
Replace Ligand is most successful when the new ligand has significant similarity to the original ligand. The new molecule is aligned with the original ligand during the replacement process, using the technique of “maximum common substructure”. This is often a good estimation of the new ligand’s docking mode. Dissimilar molecules are less likely to produce an accurate binding prediction. In the case of dissimilar ligands without obvious topological similarity, we recommend using an external docking tool to find the best starting conformation. Then import this structure for design.
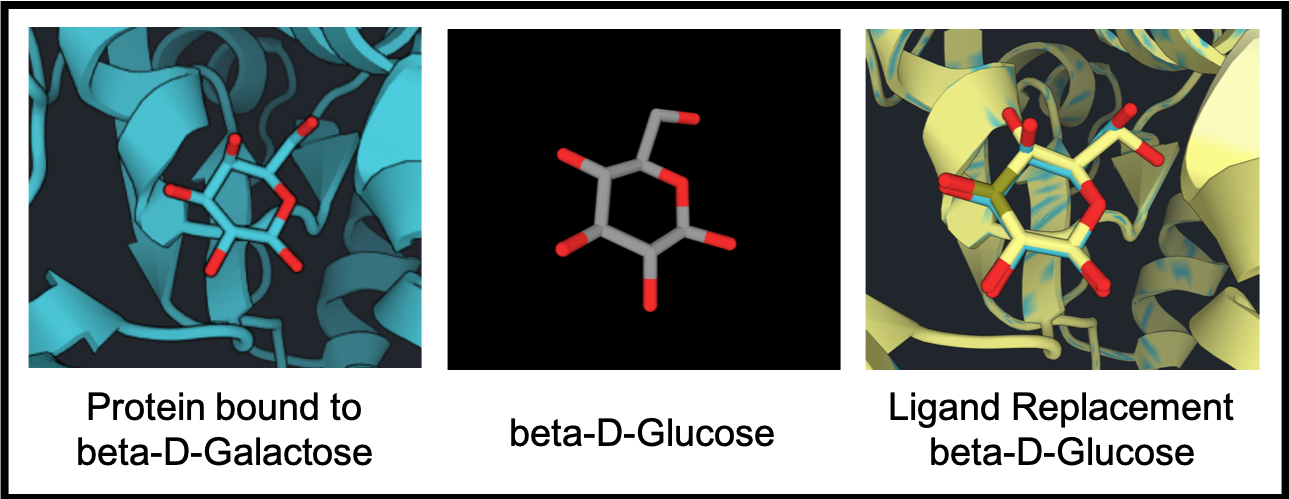
Below, a protein (blue) bound to beta-D-Galactose is used for Ligand Replacement with beta-D-Glucose (grey). After ligand replacement, the beta-D-Glucose replaces beta-D-Galactose in an orientation that is most similar to the original molecule. The new pair (yellow) is shown overlapped with the original pair.
PREPARE AND LOAD A SMALL MOLECULE
When you are loading a small molecule on its own, you need to create a sdf file. It can only have one molecule in it and it is important that the molecular model is good: With proper valencies and geometry. If your input is not good, you will get an error or a structure with abnormal features. If your structure has minor abnormalities, our software will not include the input conformation if you convert the Z axis coordinates in the file to zeros:

Our software will model up to 200 conformations, though it only displays the original structure after loading. If you convert the Z axis to zeros, the display conformation will be the most energetically favorable. However, during modeling, all conformations can be sampled in order to find one that complements the docking pose.
If you have a structure file in another format, you can convert your file to .sdf format using Openbabel. Openbabel is a free toolkit for converting chemical data between a large variety of formats. You can acquire Openbabel at: http://openbabel.org
You will convert your files to sdf either using the GUI (Windows platform) or else with the following command under Linux/Unix:
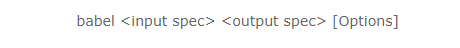
The input will be your original format (file1.xyz) and the output will be sdf (file1.sdf). Common input formats include pdb and mol2. You can learn about what options are available at: https://openbabel.org/docs/index.html
Openbable offers additional options, such as selecting a single molecule within a larger file for conversion to sdf.
iBabel is another tool which creates an easier-to-use graphical interface for Openbabel. This can be found here.
Another easy way to convert single files is available at ChemInfo.org
Once you have your sdf file ready, click  which will open a center tab. Then click
which will open a center tab. Then click  to select one or more files to be loaded. Loading will take a minute or so because we are auto-parameterizing each ligand, and then calculating a library of energetically favorable conformers for the ligand. When the process is complete, the lowest energy conformer(s) for the imported ligand(s) can be displayed by clicking on the appropriate title below the Small Molecule Loader button.
to select one or more files to be loaded. Loading will take a minute or so because we are auto-parameterizing each ligand, and then calculating a library of energetically favorable conformers for the ligand. When the process is complete, the lowest energy conformer(s) for the imported ligand(s) can be displayed by clicking on the appropriate title below the Small Molecule Loader button.
RUNNING A LIGAND REPLACEMENT
Once you have loaded the protein structure with a ligand using the Structure Loader and loaded the new ligand using the Small Molecule Loader, you are ready to do the replacement. Click the folder for the protein structure in the left panel so that the collection with your structure appears in a tab in the center window. Be sure to check the box to the left of your structure and click the  button on the right panel. This will open the Replace Ligand tab in the center window.
button on the right panel. This will open the Replace Ligand tab in the center window.
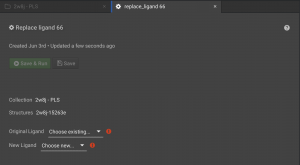
You need to select the ligand in the protein structure that you would like removed. All non-protein molecules are in chain Z. Most small molecule ligands are given a 6 character reference name. The construction of the six character name is XXXYYY. XXX is the residue sequence number of the ligand in the original input structure, while YYY is a three digital number that increases sequentially from 000 for each small molecule ligand in the system. For example, if the first ligand is the 333rd residue in the system, it will be given the CAD name 333000. If the second small molecule ligand is the 341st residue in the system, it would be given the name 341001. And so on. There are two exceptions to this numbering scheme. Monoatomic molecules are given their element as the name (rather than a three digit sequence number). So a zinc at position 335 would have a CAD name 335ZN. And water molecules are given the name HOH. So a water molecule that is the 336th residue in the system, then it is given the name 336HOH.
In this example, a small molecule occurs at position 399.
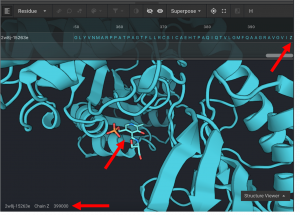
Monoatomic molecules cannot be used for Ligand Replacement at this time.
REPLACEMENT PROCEDURE
During Ligand Replacement, the new ligand is compared with original ligand in order to find an alignment that optimizes similarities between the molecules. This is done using OpenEye MCS (Maximum Common Substructure). This method incorporates Graph Theory. A molecule’s atoms and bonds make up the nodes and edges of a graph representation of the molecule. The graph aka pattern of a molecule is compared to the other molecule’s pattern in every potential orientation. These are rigid-body comparisons, not a comparison of different conformers of the molecules. Each comparison is scored by counting the number of edges (bonds) that match. A match occurs if the compared bonds have the same order and directionality. The comparison with the best score is chosen as having the maximum common substructures. If there is a tie, the atom types are also compared. If there are no matching substructures, the new ligand is randomly placed with its Center of Mass in the Center of Mass of the pocket.
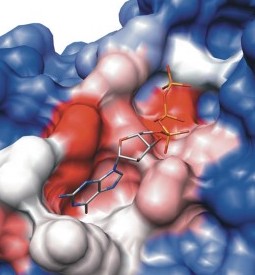
When a molecule is loaded with Small Molecule Loader, multiple favorable conformations of the ligand are created. This provides a small library of ligand conformations that are representative of the dynamic variations that the molecule can adopt. This conformation library is repeatedly sampled during modeling in order to allow the ligand to adjust its conformation to best interact with the changing protein. However, during the initial replacement process, only the first (most energetically favorable) conformation is aligned to the target.
This also assumes that the ligand that you loaded with the Small Molecule Loader is a high quality structure. This means: the structure in the loaded sdf file has near ideal bond lengths and angles and correct heavy atom valencies. This is necessary for the software to properly parameterize the molecule. If the input structure is highly abnormal, a library will not be created. In this case, the input structure conformation is used for modeling. It will be modeled in that conformation and likely have inaccurate results.
AFTER REPLACEMENT
The newly (re)placed ligand will generally need to be optimized in order to optimize its interactions with the protein. However, CAD does not do the type of extensive small molecule docking that will be necessary if the protein or ligand requires a significant amount of conformational adjustment to find an ideal conformation. An alternate docking tool is recommended if the initial orientation from Ligand Replacement is significantly far from correct. However, we have three methods for improving the interaction between the protein and the ligand. Minimize, Repack, and Relax. Since CAD has created a library of conformations for your ligand, these conformers will be sampled during optimization. This is usually necessary to find the best fit. Repack and Relax will sample conformers during modeling, but Minimize does not. We recommend doing Repack or Relax in order to sample conformers. Then following up with Minimize.
-
Minimize – This is a conservative modeling approach. This will make minor modeling changes in order to minimize Rosetta Energy for both the protein and the Ligand. However, it only looks for a local minimum. It will not sample ligand conformers, side chain rotamers, or make significant backbone changes. Though, it is great for removing minor clashes and optimizing local contacts.
-
Repack – This samples ligand conformers and side chain rotamers. It keeps the backbone rigid. This may not seem particularly aggressive, but most of the conformational improvement during modeling arises from repacking. Even small changes in a side chain or ligand can relieve major issues with a protein structure. Since this samples side chain and ligand conformers, increase the number of repeats for larger proteins and for ligands with more rotatable bonds.
-
Relax – This is the most aggressive approach. Relax runs multiple rounds of Repack and Minimize. Early rounds allow more clashing in order to sample more conformations. This is more likely to find a binding mode that differs from the original ligand, but it is also more likely to disrupt ligand interaction. So careful analysis of final ligand orientation is required.
In most instances, following up the Replace Ligand operation with a cycle of Repack and Minimize will be beneficial. In situations where the replaced ligand is very different than the new ligand, a more aggressive Relax and Minimize cycle may be required. If even Relax/Minimize is incapable of producing a suitable ligand/protein structure, it may be advisable to use an external (non Bench) docking tool to provide the initial position for the new ligand. (In instances where the replaced ligand is extremely similar to the new ligand, it may be possible to skip the Replace/Minimize or Relax/Minimize steps entirely).
EXAMPLES OF REPLACE LIGAND FOLLOWED BY MINIMIZE, REPACK, AND RELAX:
Here, we show a protein structure (2B3F.pdb) which has a bound galactose ligand. We used Ligand Replacement to insert NGA (N-Acetyl Galactosamine) in place of galactose. There are 8 protein residues that directly interact with the ligand that are shown in spheres below in each example.
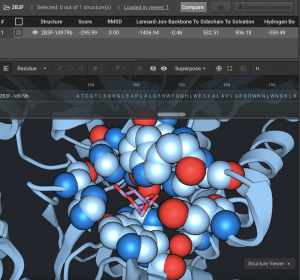
Above, you can see that all 3 post-Ligand Replacement actions shifted away from the original orientation. In this replacement, the N-Acetyl moiety was not present in the ligand before replacement so it causes a clash. Repack (bronze), changes side-chain rotamers in the pocket and rotates the N-Acetyl group. Minimize (pink), finds a local minimum by opening the pocket to relieve the clash between the protein and the N-Acetyl group. Relax (blue), makes the most significant changes. It is able to allow the ligand to pack more tightly against the protein while achieving the lowest Rosetta Energy.
You can read more about Small Molecules in CAD here.
For more information about the alignment method used in Replace Ligand, see the section on “Space State Search” from:
Horst Bunke, Pasquale Foggia, C. Guidobaldi, Carlo Sansone and Mario Vento, A Comparison of Algorithms for Maximum Common Subgraph on Randomly Connected Graphs, Lecture Notes In Computer Science (LNCS) 2396, Proceedings of the Joint IAPR International Workshop on Structural, Syntactic and Statistical Pattern Recognition, pp. 123–132, 2002.