Tutorial 4: Design Increased Affinity to a Peptide using Relax Design, Sequence Logo
Tutorial Four: Design Increased Affinity to a Peptide using Relax Design, Sequence Logo
Estimated Time: 30 minutes
This tutorial will instruct users how to use Relax Design to increase the affinity between two peptides, including how to assess whether affinity has increased. The Sequence Logo tool will also be introduced.
In order to run this tutorial, you will need to know how to use the tools from Tutorial One, Two, and Three.
(Caveat: This does not include the time it takes for jobs to run on the server. There will be several pauses while you wait 20 minutes or more for structures to run on the cloud before you can move on to more steps.)
1) Run Relax & Minimize
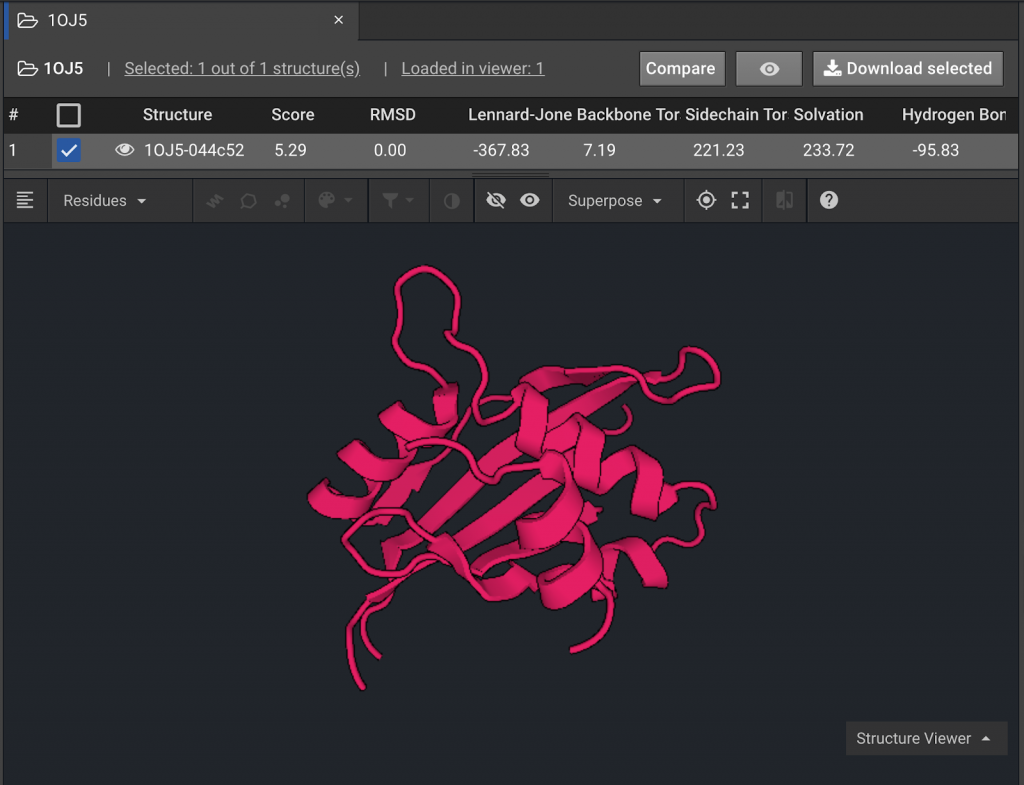
For this tutorial the structure 1OJ5 will be used to model how Design can be used to increase peptide affinity. First the 1OJ5 structure will undergo Relax and Minimize to find a starting structure with optimized energy.
Select a Project or Create a Project where you will run this tutorial. Load structure 1OJ5. Run Relax, each round with 5 repeats, until the lowest energy conformation has been reached.
Minimize this final energy structure and rename the file MinRelaxed 1OJ5. This process of Relax rounds and Minimize is described in detail in Tutorial Two.
2) Design Increased Affinity Between Protein and Peptide
To increase the affinity between the two peptide chains in the 1OJ5 structure, residues involved in protein-protein interactions will be mutated. Using Relax Design, alternate amino acids will be sampled at selected residues to increase the affinity between the two peptide chains.
Click on MinRelaxed 1OJ5 and click the  in order to visualize the protein in the Structure Viewer. One way to zoom in on the region of interest in to switch the selection mode from
in order to visualize the protein in the Structure Viewer. One way to zoom in on the region of interest in to switch the selection mode from  , which is default, to
, which is default, to  . Then click on the small peptide and click
. Then click on the small peptide and click  in order to center it. While the peptide is still selected, click
in order to center it. While the peptide is still selected, click  in order to show sticks as demonstrated below.
in order to show sticks as demonstrated below.
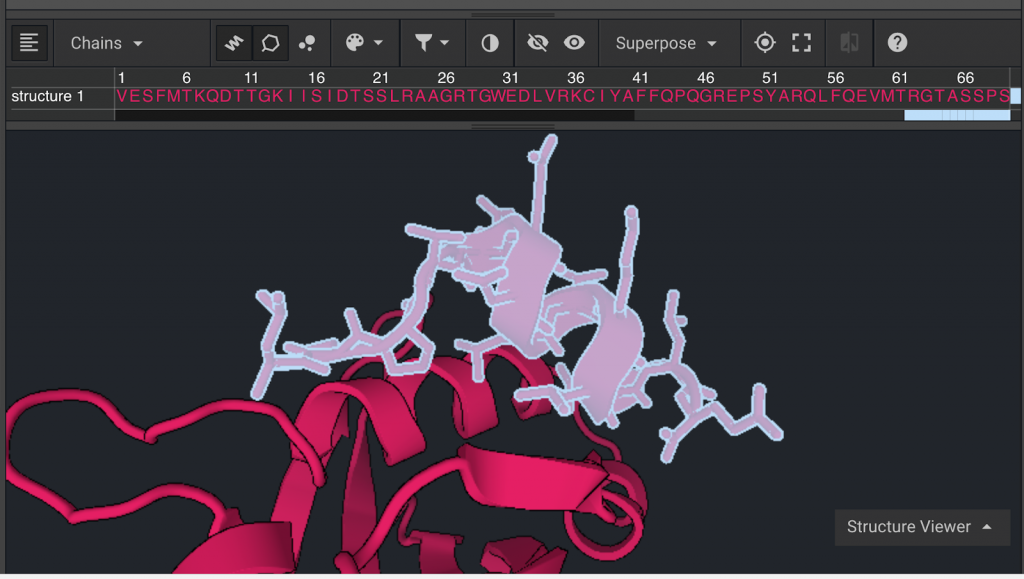
Switch the selection mode back to and select the residues whose side chains are oriented towards the protein-peptide interface. It is important to note that prolines should not be selected for mutations because it is usually disruptive to mutate away from prolines. Click  in order to show their sequences and highlight residues 106, 113, 116, and 117 (hold down command or control while selecting).
in order to show their sequences and highlight residues 106, 113, 116, and 117 (hold down command or control while selecting).
Click  and change the color in order to make it easier to see that these residues are important to binding. Create a Selector from the selection by clicking
and change the color in order to make it easier to see that these residues are important to binding. Create a Selector from the selection by clicking  then clicking
then clicking  to change the name to Peptide. Click the
to change the name to Peptide. Click the  to the left of the structure name in order to select it.
to the left of the structure name in order to select it.
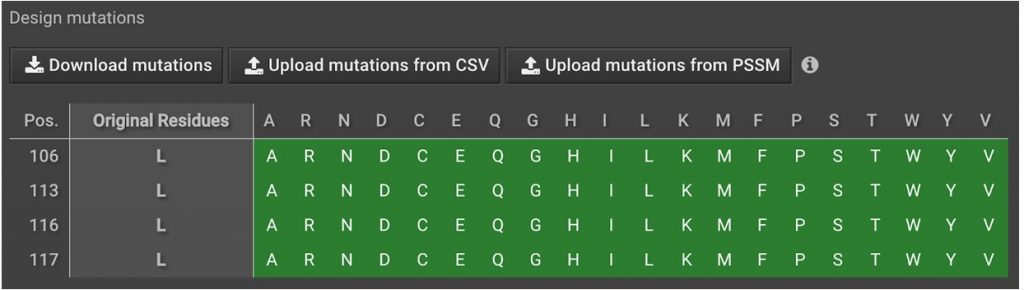
To sample alternate amino acids for the selected residues in the peptide click  in the right window. Choose
in the right window. Choose  and enter
and enter  . Then highlight all possible mutations by clicking
. Then highlight all possible mutations by clicking  in the top left corner of the mutation table as shown below.
in the top left corner of the mutation table as shown below.
Click  . Once the Design has run change the name of the Design run to relD of 1OJ5 and click on the folder in order to bring the results into the center window.
. Once the Design has run change the name of the Design run to relD of 1OJ5 and click on the folder in order to bring the results into the center window.
Click  in the lower center window and change the button to
in the lower center window and change the button to  which will display which mutations were favorable. Click the
which will display which mutations were favorable. Click the  on the top left of the Sequence Logo window and click the arrow to the right of it to select the Peptide selector. This will hide all the residues that were not part of the selector. Scroll to the right to see the four residues in the selector.
on the top left of the Sequence Logo window and click the arrow to the right of it to select the Peptide selector. This will hide all the residues that were not part of the selector. Scroll to the right to see the four residues in the selector.
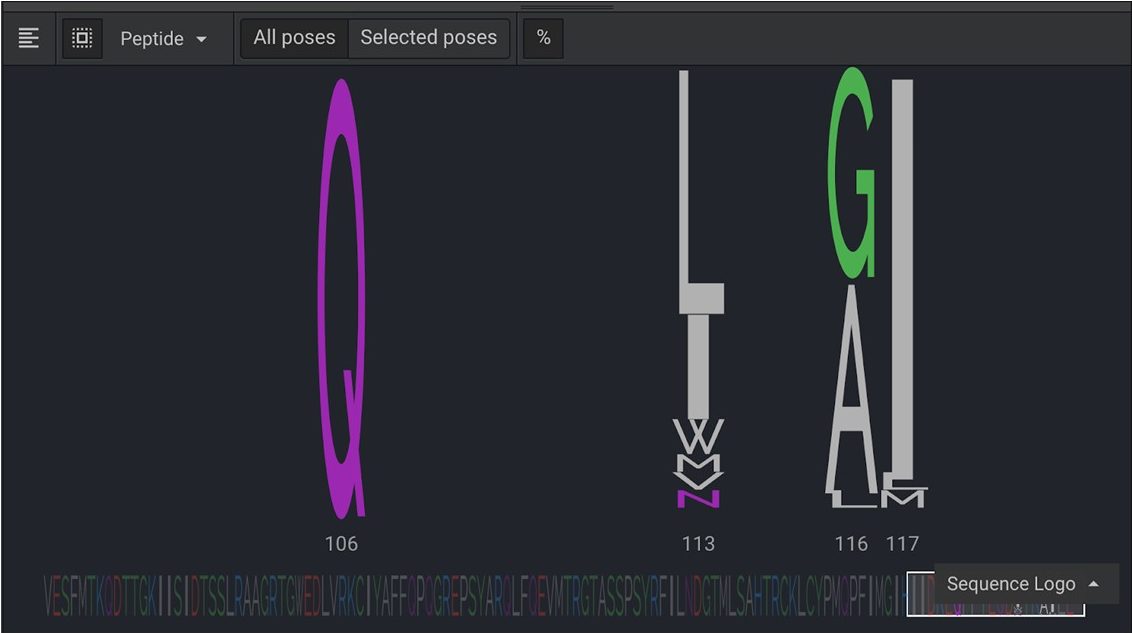
The original sequence for these four positions was LLLL. You can see that L was only favorable at position 113. Position 106 preferred Q in all of the simulations. Positions 116 and 117 favored GA and I respectively. Results above are from a larger number of repeats in order to show diversity.
To look at the structure to see why these mutations were preferred switch back to . Now, rank the structures by  and click only the
and click only the  for top 5 structures.
for top 5 structures.
Click in order to show their sequences. Then click  in order to highlight the sequence differences. Scroll over to the right to see them.
in order to highlight the sequence differences. Scroll over to the right to see them.
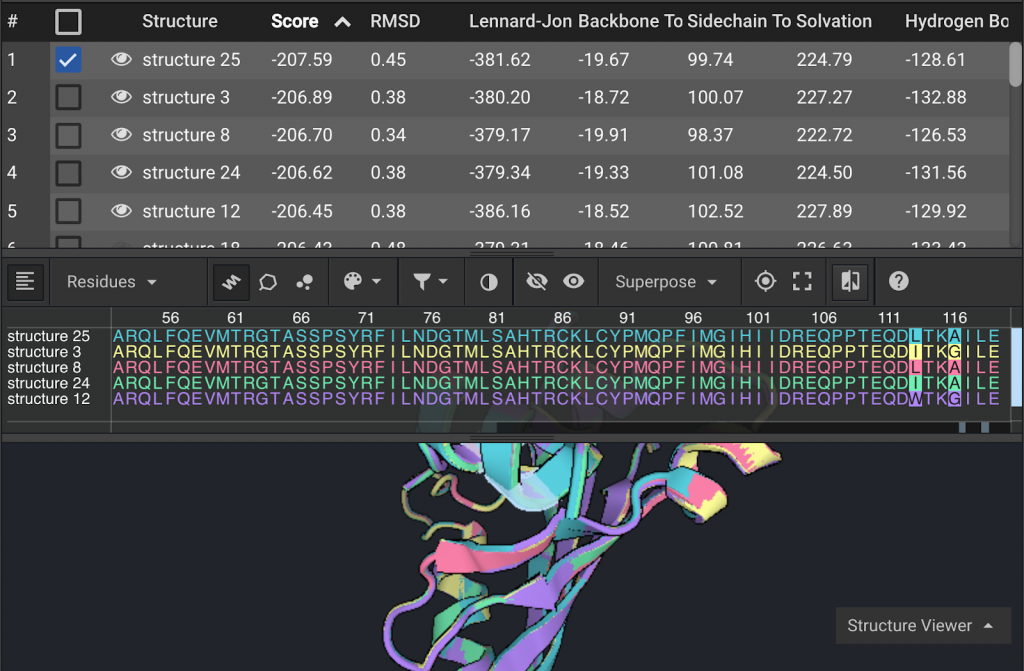
In this example there were only two positions, 113 and 116 as highlighted in the sequences above, that differed across the top five structures. As suggested in the sequence logo, residue 113 would be most stable as an isoleucine or leucine and residue 116 as alanine or glycine.
The other two new mutations which were introduced at (Q106 and I117) are present across the top five structures suggesting they are best mutations for those positions.
Viewing and Comparing Potential Mutations
The mutations which were generated with Relax Design can be viewed and visually compared to the starting residues. The lowest energy structure from the Relax Design in this example is structure 25. Deselect the for the 4 and select to show structure 25.
Click on the whole structure and click in order to show sticks. Select residues 106 to 119 by showing the sequence via , and clicking on the residues. Then click and change the color in order to make it easier to see that these residues. Shown below are the three positions that were mutated in the structure.
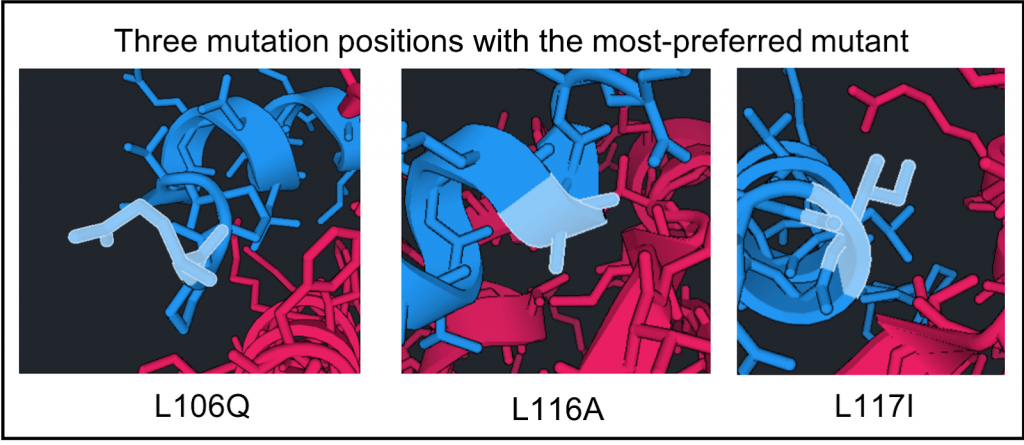
As you can see for L106Q, this side chain points away from the structure so it is probably favorable because of improvement in solvation. For 117I, the Isoleucine packs into the gap. So it probably has better packing than the Leucine did. For L116A, the smaller side chain allows the peptide to shift towards the protein’s beta sheet (see right). This may not have been sterically possible before. The fact that the other preferred mutation was a glycine supports this hypothesis.
With the L106Q mutation this side chain points away from the structure so it may be favorable because of improvement in solvation.
For L116A, the smaller side chain allows the peptide to shift towards the protein’s beta sheet (see image below).
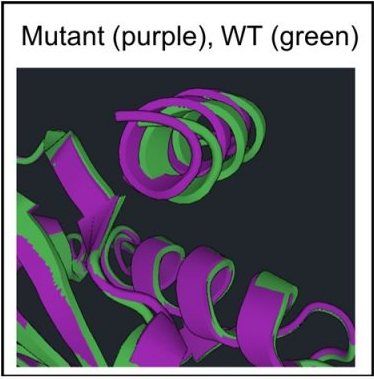
The other preferred mutation at this site, glycine, also had a smaller side chain to possibly allow the peptides to shift closer. This may not have been sterically possible before.
For L117I, the isoleucine packs into the gap which may have improved packing compared to leucine did.
In order to create an alignment of a specific region between two structures like the image in purple and green on the previous page, click  . In the drop down window, select the relaxed (relD of 1OJ5) and minimized wild type structure (MinRelaxed 1OJ5).
. In the drop down window, select the relaxed (relD of 1OJ5) and minimized wild type structure (MinRelaxed 1OJ5).
Ensure that the mutant and wild type structures are two different colors and align everything except the peptide. Do this by selecting residues 1 to 105 for both proteins. Click  to open a drop down and click
to open a drop down and click  .
.
As long as you leave the peptide out of the selection, this will align the protein so you can see how the peptide shifted when the mutations were modeled. You may want to go into full screen mode by clicking  in order to visualize the differences better.
in order to visualize the differences better.
Using Relax and Minimize to Compare Mutations
In order to verify that the mutations are energetically favorable. Click the to the left of the structure name and run a Relax with 5 repeats. Then run a Minimize on the most energetically favorable. Compare the energy of the Relaxed and Minimized mutant with the Relaxed and Minimized wild type as shown below:
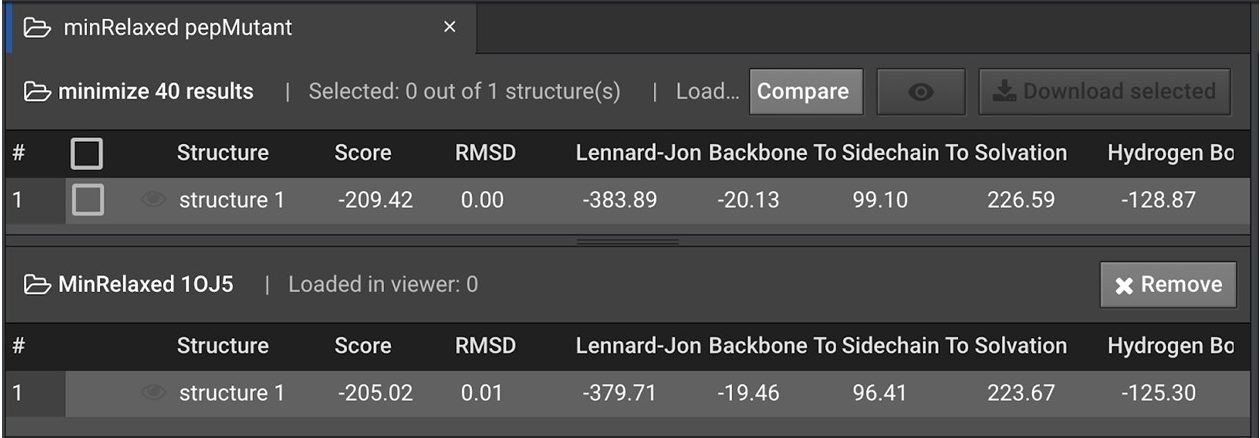
In the example above, the energy improved by 4.4 REUs from the minimized 1OJ5 structure to the minimized Design result. This is a significant difference for only 3 point mutations. However without testing, it is not clear that the mutations have improved affinity. The mutations with lower energy structures may have improved stability within the peptide which doesn’t affect affinity.
Predicting Peptide Stability Versus Peptide Affinity
One way to computationally predict whether the mutations are favorable due to peptide stability is to model the peptide without the protein. You will need to download the structure for 1OJ5 with only the smaller peptide chain present.
You can manually delete selections of the protein in a text editor or you can use a visualization program such as Pymol. The manipulated file needs to be a .pdb file with only the peptide residues present.
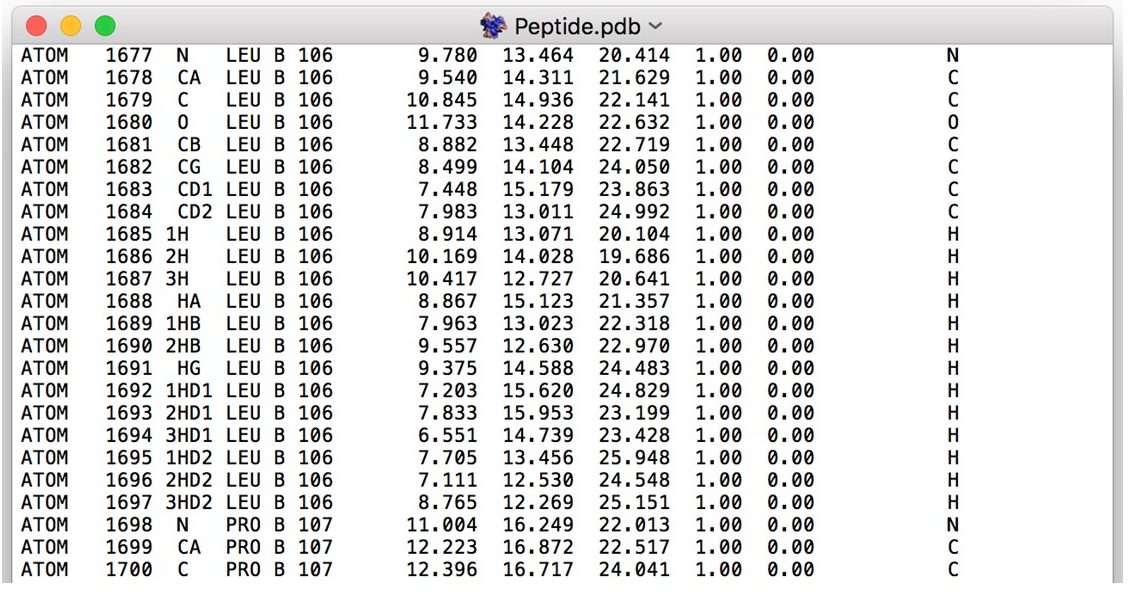
To download the original PDB file click the 1OJ5 folder to bring it into the center window. Click the to the left of the structure name in order to select it. Then click  to open a drop down window that allows you to select pdb, fasta, or scores. Select PDB Files. Once only the smaller peptide chain residues remain the PDB file should look like this:
to open a drop down window that allows you to select pdb, fasta, or scores. Select PDB Files. Once only the smaller peptide chain residues remain the PDB file should look like this:
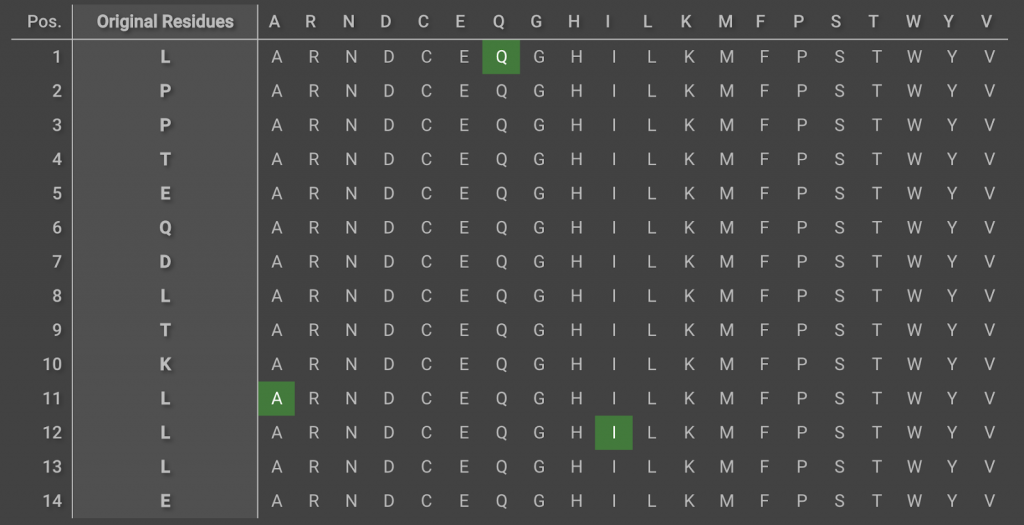
Load this new structure into CAD as a new project. Create the three point mutations by running a single  run, noting that the position numbers have been renumbered. You do not need to create a Selector for this. You can select the mutations manually in the mutation chart of the Design tab. Create these mutations:
run, noting that the position numbers have been renumbered. You do not need to create a Selector for this. You can select the mutations manually in the mutation chart of the Design tab. Create these mutations:
Leave the number of repeats as 1 and click . Rename the folder Peptide Mutant. The WT Peptide and this new Peptide Mutant should be Relaxed and Minimized before comparing their energies. Since the peptide is so small, running 5  repeats is more than enough for both the WT and the new mutant. Then run
repeats is more than enough for both the WT and the new mutant. Then run  on the one with the best energy for both the WT and the new mutant. Using these, click to see the energy difference.
on the one with the best energy for both the WT and the new mutant. Using these, click to see the energy difference.
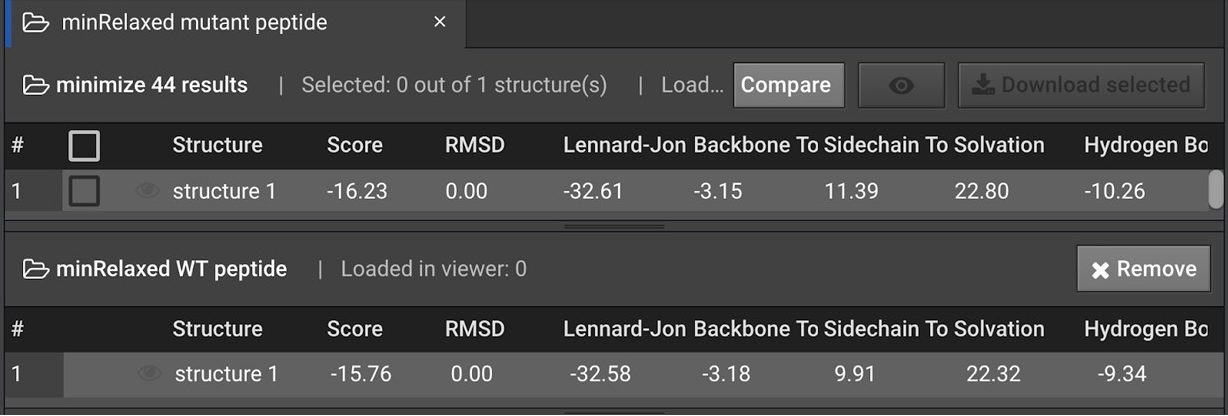
In this case, the energy improved by 0.5 REUs, suggesting that the mutants are more likely to affect affinity to the protein than inner stability.