Frequently Asked Questions
Is it possible to run Repack on a region of a protein?
Currently CAD does not include a version of Repack that can be limited to a single region; Repack will always include the entire protein. However, this is on our list of features to eventually add to CAD.
Is there a limit on the size of a structure I can model through Homology?
Yes, there is a minimum of 50 residues and a suggested maximum of 1000 residues for Homology Modeling runs. In order to model larger structures, we suggest separating domains, e.g. through BLAST, and running these as individual targets.
Run time will range from a fraction of an hour for smaller structures, up to ~12 hours for 800 residues.
How are template weighs calculated in Homology Modeling?
In Homology Modeling, each alignment is given a likelihood score. This score attempts to calculate the probability that a particular template/alignment is among the top 2% GDT (Global Distance Test, Zemla et al., Proteins, 1999) score for a particular target. The likelihood measure only considers two factors when assigning a value: the generation method, and the method’s ranking of the alignment. The values used were determined by evaluating the performance of each alignment method on a large benchmark set of 1,431 targets (Song et al., Structure, 2013); a function of the following form was fit to this data:
Here, mthd is the alignment method used, and rank is the individual method’s ranking of the alignment.
How can I tell if a designed sequence is a good one?
This is a very common question. In general there are two ways in Bench to tell whether a sequence is good: 1) By checking the structure’s score and other metrics in the output of a design: overall lower scores are better and 2) By computational testing with Repack, Relax and Bench Homology. These two approaches are explored in detail below:
1) SCORE METRICS
A straightforward way to see whether a structure has improved after running Design is to look at the structures’ scores. If the score is lower after running Design that is a good indicator that the overall protein structure has improved.
Some protocols will allow partial scores to be calculated — e.g. just at a protein/ligand interface or at a protein/protein interface instead of the entire protein. Lower partial scores are very frequently used to identify the best output sequences and to figure out when a design is improving.
If you are designing for overall stability, then total score is a good metric. If you are designing for a certain function, then a combination of the best partial score and the improved total score should be used to assess the structure.
2) STRUCTURE COMPUTATIONAL STABILITY METRICS
A designed protein sequence really consists of a sequence and its full-atom structure. A good sequence is one where the sequence and structure are at an energy minimum, in other words where the designed structure is bio-physically stable in solution. Cyrus Bench contains many methods to test stability of a structure: Repack & Minimize, Relax, and Homology (not in CAD).
The type of stability testing you should do to test a design depends on how much you have changed the sequence and structure, as follows:
Test A: Small number of mutations (fewer than 10) on a fixed backbone or only lightly minimized backbone
- Run Repack & Minimize on the final designs that you want to evaluate. If the side chains shift significantly, especially in the protein core or near the core, then the designs are not as good. However side chains on the surface may shift, especially if they have few contacts.
Test B: More aggressive design of one stretch (e.g. 4 or more mutations in a row), or significant (more than 0.1 Ångstrom) changes to backbone
- In this aggressive design scenario if the results pass the test in (A), also try running Relax. If the structures after running Relax drifts less than 1 Angstrom in the designed region, these are good designs. If they drifts less than 0.5 Angstroms these are very good designs. If it drifts more than 1.5 Angstroms, you may consider further redesign. For example you may consider using the output from Relax as input for the next stage of design.
Test C: Mutations to more than 15-20% of residues in the protein or designed (intended) backbone changes of more than 1-2 Angstroms
- These can be tested in (B), and it is very positive sign if they are passing in (B). For an even more stringent test consider running these as input in Cyrus Bench Homology. This assumes that you have sequence homologs, which you will by definition if you began your design from a published structure in the PDB or a privately held structure.
- If the structures are coming back to within 2 Ångstroms especially in the designed region, then these are very high quality and self-consistent designs. Achieving this type of design can require multiple cycles of design and homology modeling to converge on a sequence/structure pair.
How many repeats should I run?
For most CAD modeling tools, a single run will be insufficient at sampling the variety of protein conformations. Each run begins with its own unique trajectory and one run will have a slightly different answer from another run.
The more possible conformations your protein may have, for example if it is especially flexible or large, the more repeats will be required in order to fully sample the possibilities. Additionally if you believe that your final structure will be significantly different than your starting structure, more repeats will be required.
For most cases, 100 repeats will be more than sufficient. Once you have completed a run with 100 repeats you can check whether 100 repeats is sufficient by looking at the at the range of structures’ scores, which corresponds to the energy of the structure. For an easier view of the range of structures’ scores use the Scatter Plot to view the scores of all 100 structures.
- If the range of scores is greater than 50 Rosetta Energy Units (REUs), use the lowest energy structure from that run as the input for another run this time with 200 repeats.
- If the energy range is greater than 100 or 200 REUs, use the lowest score structure as the input for another run with 500 or 1,000 repeats respectively.
Once you have fully sampled the search space, the scores for the structures’ will stop improving. Continue repeating iterative rounds of repeats as detailed above to reach this point.
Reasons to increase the number of repeats are case-specific…
- Repack – Samples side-chain orientations. Increase repeats for large, flexible proteins.
- Minimize – Only 1 run is necessary because this method differs than other sampling method. It is deterministic and will only find one answer no matter how many times you run it.
- Relax – Samples global structural changes. Increase repeats for large, flexible proteins.
- Loop Rebuild – Samples loop structure. Upper limit is 12 residues. Increase repeats when running Aggressive mode.
PROTEIN DESIGN METHODS:
- Design – Samples side chain orientation and allows mutation.
- Flex Design – Samples global side chain orientation, samples backbone near area of design.
-
Relax Design – Samples global side chain and backbone and allows mutation.
- Increase repeats for large, flexible proteins.
- Increase repeats when more permutations are possible.
- Allow more repeats for Flex Design than Design. And more repeats for Relax Design than Flex Design.
HOW TO CALCULATE NUMBER OF PERMUTATIONS:
Example – For 5 mutations position that are all allowed to sample all 20 amino acids,
- The number of permutations = 20 x 20 x 20 x 20 x 20 = 3.2 Million.
- Number or repeats needed = # of permutations / 50 = 64k.
It is not recommended to run that many repeats, so we recommend splitting the job into 2 runs:
- The first run has 20 x 20 = 400 permutations, 8 repeats.
- The second run has 20 x 20 x 20 = 8,000 permutations, 160 repeats.
- Then use the preferred mutations from each run to make a final design at all 5 positions
What is the next step to best optimize Design after I have run Prepare on a structure? Do I go straight to Design, or should I run other actions?
Prepare has been tuned to optimally minimize the energy of a structure so that it is ready for Design. It should not require any subsequent Repack/Minimize/Relax before going into Design.
After Prepare, you can go ahead and run Design with the following number of repeats:
- 10-20 repeats for a short sequence of less than 10 residues.
- 50-100 repeats for a larger protein, or up to 200 if you are searching for more diversity in outputs.
After running Design, you may find that your sequences are all very similar or that they are fairly diverse. The Sequence Logo in Bench and the Compare tool may be helpful when analyzing the diversity of Design results.
Depending on your goal you may want diverse structures with a variety of mutations or you may want less diverse structures with a smaller variety of mutations.
If you are not getting sufficient diversity, you will need to take a more aggressive approach through any of the following options:
1.) Run Prepare again, or run it 10-50 times to get more structures. Prepare is stochastic; each output will be slightly different and will give diversity in design.
2.) Run Minimize and then Relax on a number of structures (e.g. 10-20) and then use those outputs as input for Design. Again, this will encourage diversity in designs.
3.) Most aggressively, you may perform a Relax on your structure (or many Relax runs), pick the ones that don’t drift too far (low RMSD in the filter panel) and then run Design on those. This is a fairly aggressive approach, and will give you a immense diversity, but this approach is useful if you would like to identify many different mutations.
How many mutations can/should I make at a time?
Although Cyrus offers best-in-class protein design, there are limits to what it is recommended to attempt. As seen in this question it is possible to redesign 20% or more of a sequence, but it requires many cycles with separate tools, not a single run through Design. This is an inherently hard experiment that Cyrus Biotechnology has made easy to perform: be careful that the ease of selecting mutations does not tempt you to try too many at a time.
The red flags that your design problem requires more computational confirmation (detailed above) are if:
-
You are designing more than a handful of point mutations
-
You are designing neighboring residues (in either sequence or Cartesian space)
The most conservative route would be to break your design problem into as many small Design operations as possible, verifying their success in-between with the other Bench tools.
How can I pull out the sequences I generated with my Design?
You can export the sequences generated in Design by downloading the structures’ PDBs or FASTAs. Begin by opening the file containing your Design results.
As shown below you will be able to select the structures you would like to download data for by checking the box next to each structure’s name. If you would like to download data for all the structures in the collection select the top most box.
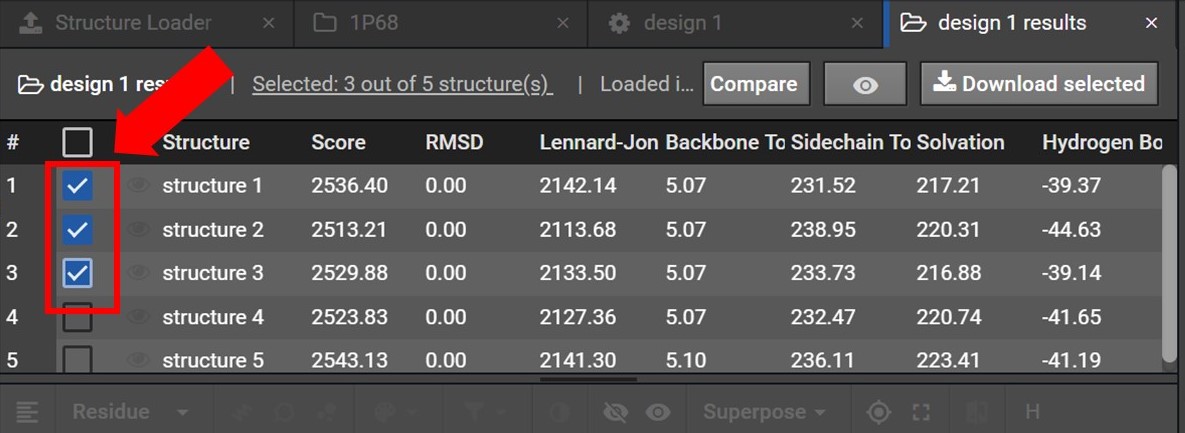
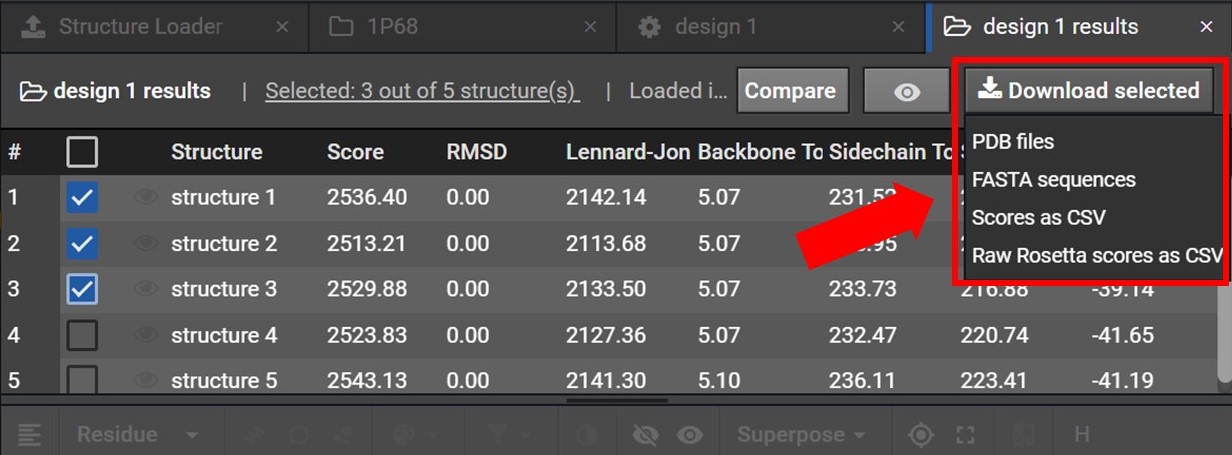
Once your structures have been selected click  , located at the top right. As shown below this will open a drop down menu where you can choose to download either the PDB files or FASTA sequences for these structures.
, located at the top right. As shown below this will open a drop down menu where you can choose to download either the PDB files or FASTA sequences for these structures.
Additional information on how to analyze the FASTAs from Design results can be found here.
How should I decide how many repetitions to run and what combination of Actions to do?
The number of repeats and the unique combination of Actions you should run when making mutations to your protein will depend on two factors. First, how large will your following experimental tests be; how many sequences are you looking to generate? Second, what level of protein optimization are you working at; how many mutations do you intend to make? Consider which parameters and scenarios below are the best fit for you:
SIZE OF EXPERIMENTAL TEST:
- Small: a few sequences
- Medium: hundreds of sequences
- Large: thousands or millions of sequences
LEVEL OF OPTIMIZATION:
-
Point mutations: You are looking for small changes, just single point mutations in each tested sequence. For example when the protein you are working with is fairly well behaved or already performs well in your assay.
-
Multiple mutations: You are looking for many changes, multiple mutations at a time per tested gene sequence. For example when the protein you are working with has little or no activity.
Below are several recommended courses of action based on the parameters described above:
1.) Small test for point mutations:
For a small sized test of point mutations use the DDG tool. Instead of using the DDG tool directly you may also run Design first with a very tight filter on total score to identify mutations and then test these mutations in DDG. For information on how to use DDG click here.
You may need to download the Design results into a sequence viewing tool to identify the common mutations. For additional workflows on analyzing sequences from Design results click here.
2.) Large test for many mutations (library design):
For a large sized test of many mutations alternate between runs of Design and Minimize to generate many sequences in batches. These sequences can then be downloaded collection by collection for sequence analysis to identify common mutations at each position. These can then be generated by degenerate codons to create large library sizes.
3.) Small test for many mutations:
For a small sized test of many mutations alternate between runs of Design and Minimize to generate greater sequence diversity. Then you may consider two different approaches. One option is to filter structures stringently by total score and only select a few of the best scored sequences to order.
A second option is to download many of the sequences and generate a phylogenetic tree in a sequence analysis package. This will allow you to pick the most “representative” sequence from each branch of the tree. This approach is a way to sample lots of diversity but only test a relatively small number of sequences.
Is there a way to narrow down Designs so a few models can be screened for retained activity? Is there a way to run multiple DDG mutations together?
A common question is how to pick the best Designs among the ones (already likely good energetically) that Rosetta produces.
One helpful step is to look at the multiple sequence alignments of outputs and build a phylogenetic tree to understand the diversity over your set, then tune your choices as described there. This is also relevant to which mutations to accept when redesigning a protein, especially just using Repack to test your design’s stability in Bench.
DDG was calibrated empirically on thousands of real mutational data sets of single point mutations — so it gives one free energy number for one mutation, and in our testing that free energy correlates well with experimental data. In theory one could also do such a thing for double mutations, as described in this workflow, where we have some good evidence that it works, but for situations beyond that the theoretical assumptions of DDG start to be less valid (e.g. assuming fairly rigid backbone) and we don’t have good benchmarking data to rigorously support its use, the sequence space is just too high.
For more information on how to decide which combinations of Actions and repeats may be best for your project, click here.
Can loops be modeled in Homology Modeling and then inserted in an existing structure in CAD?
Cyrus Bench Loop Rebuild is currently in Beta testing. However, the method proposed is great and routinely used to do loop extensions. Here is a suggested workflow.
- Insert desired loop sequences into your FASTA sequence, then run in Homology Modeling as a structure prediction.
- Load the resulting structure into CAD.
- Optionally, you can “test” a structure by running some Relax (say 5 runs) to see if the new loop is stable. If it is, that’s a great sign.
- If not, you may run 1 –> 2 –> 2a with some sequence ideas until you get a loop structure that looks right.
- Run one or two cycles of Design/Minimize (e.g. Design/Minimize/Design/Minimize) on the loop alone, or the loop and its neighbors.
- Test the final loop with Relax again to make sure it does not fall apart.
- This can be done iteratively, e.g. go up to 3, then take that design sequence back to step 1, and look for convergence of designed sequence to intended structure.
A “good” new loop is one that has a sequence/structure combo that is stable to Relax.
What is considered good change in ddG when looking for stabilizing mutations?
When looking for stabilizing mutations it is helpful to compare DDG results across different models. The standard deviation is normally 1-2 between models, so a DDG of less than -1 would be interesting to look at.
For more information on how to run DDG and interpret results click here.
I don’t see disulfide bonds in the viewer. Are they being included in Bench calculations?
The Structure Viewer used in CAD and HM does not have a way to visualize disulfide bonds, but they will still be accounted for by Rosetta during any calculations.
We are planning on switching viewers in the future to one that will allow for disulfide bond visualization.
What quality of structure do I need as input?
In order for Bench to give useful results, the input structure does need to be accurate.
-
For Good Results: Crystal structures should ideally have resolution at or better than 2 Ångstroms.
-
For Usable Results: Crystal structures with a resolution of approximately3.5 Ångstroms are the low end of usable results.
-
Non-crystal structures (NMR models) tend to perform somewhat worse.
Additionally, if Relax models of your starting structure (generated by running Relax only, not with Prepare) are converging, then that is a good sign that you can continue with your modeling experiment.
If your crystal structure has a well-defined core but missing loops, you may wish to try the Homology Modeling tool, ensuring that your existing structure is available as one of its inputs. This may be able to re-mediate problems in the loops.
For more information on how to use the Homology Modeling tool click here.
What is the recommended browser for use with Bench?
Cyrus products were built to run on Google Chrome. If you are unable to use Chrome, Firefox is your best alternative.
Which Rosetta version and flags are used on a Bench run?
The version of Rosetta underlying Cyrus Bench CAD is e3f2cae19743fcd2ee7452da25072bb3b4a639f9, which dates to April 27, 2017.
-
Exception: The version of Rosetta that the DDG metric is currently running is
981ca1ab37cf1092da719de54cee4cfd8a73867e, released on July 23, 2015 -
DDG is running another version because our most complete and thorough benchmarking results indicate that specific version has performed optimally, and newer versions have not been sufficiently tested to positively affirm their scientific quality for this specific application.
-
The version of Rosetta underlying Cyrus Bench HM is
6d43786b5bf9572f7da090dff963244eb31ee1aa, which dates to November 23, 2016.
ROSETTA COMMANDS AND FLAGS:
- Prepare = Relax with these extra flags
- relax:constrain_relax_to_start_coords
- relax:coord_constrain_sidechains
- relax:ramp_constraints false
- Repack = Fixbb with flag
- repack_only
- Minimize = Minimize
- Relax = Fastrelax
- Loop Rebuild = Loopmodel (aka Next Generation KIC)
- loops:remodel perturb_kic
- loops:refine refine_kic
- kic_rama2b
- loops:ramp_fa_rep
- loops:ramp_rama
- kic_omega_sampling
- allow_omega_move true
- Design = Fixbb
- FlexDesign = Coupled_moves
- RelaxDesign = Fastdesign
- Epitope Scan = Rosetta_Scripts described here
- DDG = Ddg_monomer
- Antibody HM = RosettaAntibody3 with flags:
- – name: num-out-models
- value: 10
- – name: exclude-homologs
- value: false
- – name: no-relax
- value: false
- – name: output-ab-scheme
- value: Chothia_Scheme
- – name: num-out-models
GENERAL FLAGS:
- SCORE flag:
- score:weights talaris2013
- LIGAND flags:
- ignore_unrecognized_res false
- ignore_waters false
- load_PDB_components false
- PACKING flags:
- ex1
- ex2
- extrachi_cutoff 0
- use_input_sc
- ROTAMER flag:
- relax::limit_aroma_chi2 true
What should I use to view alignment JSON files?
We are currently working on displaying the alignment in the structure viewer in Homology, but currently the best option is to read the alignment JSON file in a text editor that supports monospaced font (we use Atom). Once you’ve made sure that the alignments are in a fixed-width font, copying/pasting them on consecutive lines should allow for comparison.
Can I kill a Homology Modeling job after it has been submitted?
No, if you would like to abort a job email support and we will stop it. We are looking into allowing users to stop Homology Modeling runs before significant computation has been spent on the job.
How are metals taken into consideration in Homology?
During the simulation the metal ions are removed however HM has constraints to the backbone of the templates that helps to preserve the open/close conformations.
Once we implement template selection you will be able to select the templates that match the conformation you are interested in. We do plan to add full metal support to the pipeline. The way we envision this is by letting you select the metal ions from one of the templates that you want to go into the simulation.
How does credit accounting work?
All Actions, Structure Predictions, and Analysis tools consume credits.
Anything that uses credits is activated with a green  button. Though the button can say Run, Save and Run, or Run x Sequences depending on which tool is being used.
button. Though the button can say Run, Save and Run, or Run x Sequences depending on which tool is being used.
Warning: Once you have clicked the green button, you cannot stop the job by deleting it. Deleting jobs will only remove them from view.
For certain applications that consume considerable cloud resources, you will be alerted to credit usage before the job is started.
How can I view my credit usage history?
You can verify the amount of credits you have used by going to the CAD homepage and clicking the Accounts link on the left. Then click Download History as shown in the image below.
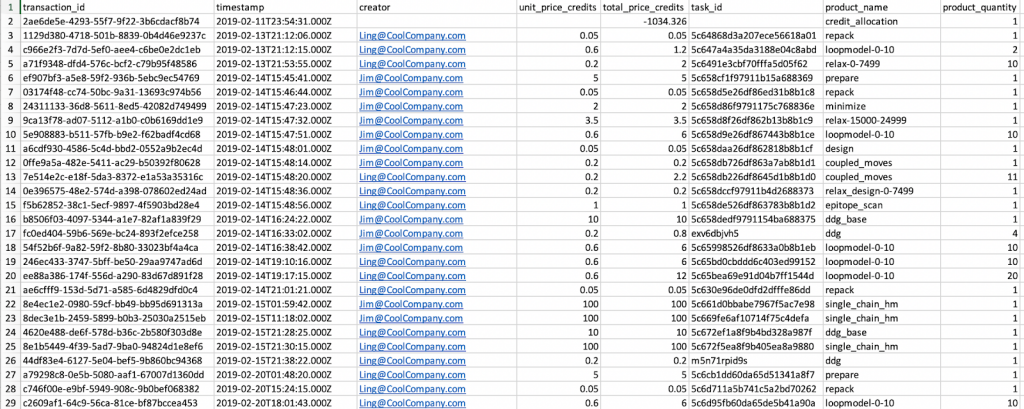
This will download transactions.csv which will list all your transactions, as the name implies. As shown below it will list transactions for the entire company on the account, but you can sort the list by the “creator” column if you want to see jobs run by a single user.
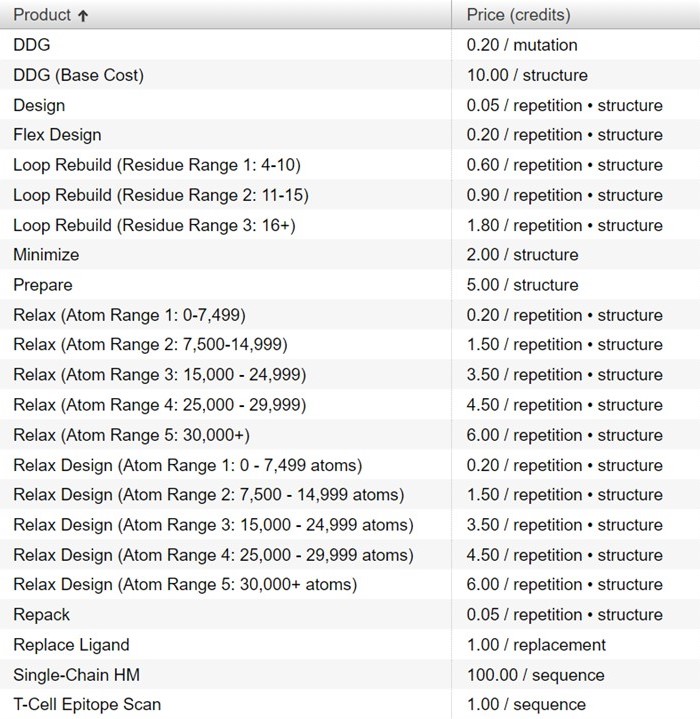
How many credits does each action in Bench take to run?
The complete list of product costs are available under the Catalog tab in Bench and are shown below:
How can Bench be used for my custom project?
Cyrus Bench is meant to be provide an easy-to-use GUI so you can run Rosetta without the months of necessary training that are needed for the full version of Rosetta. Bench has the majority of Rosetta tools that are needed for most structure prediction, modeling or design jobs.
Additional Rosetta tools are often needed for more complex research goals. At Cyrus we offer a wide range of Fee-for-Service jobs that we can run for you manually and quickly, by expert hands. We will work with you to help make Bench more useful for all your protein modeling needs.
Please email support@levitate.bio to ask about additional modeling that you may need.
We commonly help with these simple tasks that often use a modest number of credits:
- Homology Modeling
- with custom templates
- with ligands (small molecules, DNA, RNA),
- with protein-protein complexes.
- Protein Protein Docking
- One on one consultation with a Rosetta modeling expert
We also can help with larger projects such as:
- Automated De-immunization to remove T cell epitopes
- CryoEM map refinement
- Cyclic Peptide design
- Modeling of Non-canonical residues And so much more. Please schedule a call so we can discuss any additional meeds that we can help with support@levitate.bio.
# Modeling with DNA/RNA
Cyrus Bench has implemented DNA/RNA handling in order to allow loading and modeling with these highly biologically-relevant molecules. Rosetta is excellent at modeling with nucleotides present. It has been successful at modeling proteins with DNA and RNA for deriving affinity, specificity, and for exploring the effects of mutations. However, there are a few limitations that you should be aware of before working with DNA/RNA in CAD.
Warning: Our Structure Viewer has an issue that sometimes makes DNA/RNA look broken. To verify that an apparent break is not being modeled as a break, download your structure from CAD and open in another viewer such as pymol. If the DNA/RNA is not broken in this viewer, then you can be certain that the model is not modeled as broken. This is important because there are some non-canonical DNA/RNA types that will break and some that will just look broken.
Also, DNA and RNA are modeled as rigid-body structures. Rosetta would need additional score terms in order to model DNA flexibility properly. And RNA modeling is one of the most challenging things to do with Rosetta. So, we recommend partnering with an RNA specialist if you want RNA flexibility. For most modeling cases, rigid DNA/RNA is sufficient. Contact us if you would like further advice at support@levitate.bio.
LIMITATIONS
Our DNA/RNA modeling is new code. So this early version is not a comprehensive tool. The current version is mostly limited to canonical DNA/RNA.
There are many non-canonical DNA/RNAs that we will eventually be able to handle, but this early version is limited to A,U,T,G,C (for the most part). Your non-canonical residues will be initially deleted from the structure when you have a structure in Structure Loader. You can select the non-canonicals and undelete them before accepting changes and allowing everything to be loaded into CAD. However, you should verify that the residues are correct by downloading the file from CAD and manually inspecting the residues in another structure viewer. Our Structure Viewer has a known issue with DNA and RNA that makes some regions appear broken even though they are normal. Things that look broken in the Viewer will model normally.
So, some non-canonical DNAs will undelete and load into CAD normally. This should be verified because our Viewer is unreliable at viewing DNA at the point. This is an issue we are working hard to improve.
Non-ideal DNA/RNA structures will lose important features or not load at all
Many structures in the PDB or from other sources will have bond lengths or angles that are too far from the norm so will not be recognized properly. Commonly, the offending nucleotide will be treated as a separate small molecule than the DNA/RNA that it is connected to. Our Structure Loader will drop its covalent connection to up and downstream DNA/RNA and create a small molecule out of the remaining atoms. This would likely cause significant problems if you tried to model with this inaccurate variation, so you can either delete the small molecule or fix the input file. Feel free to email us the file so we can try to fix the molecule for you.
Example: PDB ID 6D0M: DNA Polymerase eta bound to DNA/RNA and Cytarabine
Above, you can see 6DOM in our Structure Loader. One nucleotide in chain T position 1 labeled DC has an X to the right. This indicates that the atoms are too far from ideal to be included in the structure so have been deleted. Below that are two nucleotides in chain P positions 1 and 2 labeled DA and DG. These had non-ideal atoms, but not so bad as to be deleted so they were modified in some way. To get more information, hover your mouse over the position on the structure or over its name to the right or above.
In the above example, DG at chain P position 2 had warning: altLocs: A, B. This means that during processing for this residue, it could place an atom in the residue at 2 locations, A and B. It discarded option B.
In the case of DA at chain P position 1, the warning reads the same, but you can see that it loses its covalent attachment to position 2 and is handled as a separate small molecule (below).
Again, DA at P1 gets the altLocs warning indicating non-Ideal atoms, but it has an additional problem that does not include a warning. You may note that the cartoon mode appears empty. This is a Viewer Error that occur for some cases. Above center, this structure has been loaded with all molecules accepted. Then it was opened in the default Structure Viewer which has more features than the Viewer available during Structure Loading. Here you can clearly see the molecule in sticks mode and it does not appear to be covalently attached. It only shows the adenine bound to pentose as if it was converted to a nucleoside. This is just a Viewer Error. If you download the file off CAD, you can see that the normal DNA is present. And more importantly, it models normally. We just need to fix the Viewer Error.
DNA/RNA will not work with DDG or Repack
Normally, you have the option to turn on or off Repack for any residue or small molecule present in your structure. This is because side-chains and small molecules have a set of alternative conformations that can be sampled during modeling. However, DNA and RNA are not handled this way. During modeling, the original conformation is held rigid. The protein and other molecules are allowed to move in relation to DNA/RNA, but DNA/RNA does not change.
Our DDG tool does not run with any non-protein molecule present. This tool may enable it in the future, but it needs to be benchmarked to see how predictive the tool will be with small molecules and/or with DNA/RNA. This is something we are working on, but cannot promise that DDG will be a good tool when small molecules are present. So, in the event that we find it effective, we will work on adding this feature in CAD.
How do I add a new user?
Please email support@levitate.bio to request to add another user.
What are valid names for collection?
If you see this error:
Please ensure that your collection name contains only valid characters as defined at levitate.bio/support
You have entered a non-accepted symbol into a name field. Here are the only symbols that are allowed in name fields: :.\-_&+()%,
How can I reset my password?
You can reset your password by visiting cad.levitate.bio and clicking “Don’t remember your password?”.
How do I create a bug report?
If you are experiencing unusual behavior is CAD, we will be happy to help you fix the issue.
Some issues can be resolved by closing all tabs, exiting CAD, and then reopening CAD. Some issues will disappear during this process because they are browser-related (we recommend Chrome). If that doesn’t work, please send us a bug report.
We appreciate as much detail as possible about the behavior in order for us to diagnose the problem. We would like you to email us the description of the behavior, a screenshot or shots of CAD highlighting the involved windows, and some debug information from the Chrome Browser.
1.) Take one or more screenshots of CAD. Click on the task that causes the unusual behavior in order for it to appear in the center window. Take a shot that includes all CAD windows in order for us to see the status of the task in all three windows.
Mac – Click Opt-Command-4, then click and drag the region of interest. When you release the click it will save a screen shot to the desktop.
PC – Click Ctrl-Print Screen to copy the entire screen. You can paste it into a text editor or paint.
2.) While you have the job that causes the odd behavior selected, click View in the Chrome task bar on top. Then click Developer and Developer Tools.
This will open a console with debugging information. Copy all the test in the console and paste it into the email for us to analyze.
3.) Send the email to support@levitate.bio. Describe the behavior. Paste the debugging info from the Developer console. Attach the screenshots.
4.) Wait for a response email from support that should arrive within one business day. Hopefully we will have a fix for you immediately, but if not, we will start working on the problem immediately. Any information you can add will help us diagnose the problem.
The sequence numbers upon structure import don’t match the PDB file
When a structure is imported through the Structure Loader, Bench concatenates all chains in the PDB file into a single long string of residues. The chains will be in the same order as they appear in the PDB file. This reflects the protocol used by Rosetta.
As a result the numbering of residues in chains beyond the first will not match that from the input PDB file. Each residue number in the second chain will be increased by the number of residues in the first chain. Each residue number in the third chain will be increased by the sum of the numbers of residues in the first two chains. And so on.
We will provide the ability to retain your original numbering in a later update of Bench.
What are the scoring metrics in Bench?
The scoring metrics from HM and CAD are listed below as they appear left to right (lower is better):
- Score is a summary of the Rosetta energy function, reported in Rosetta Energy Units. When no other specific considerations apply, this is how one should rank models. This score includes entropic and enthalpic components, statistical terms and physical terms. It will not necessarily equal the summation of the other presented terms (minor terms are not displayed).
- Lennard-Jones measures how “well packed” a protein is by using a Lennard-Jones potentials to score atomic attraction and repulsion. A positive score here indicates clashes, implying some problem in the model. A negative score is expected.
- Backbone Torsion quantifies how realistic the backbone angles are, using the Ramachandran plot. As a probabilistic score, this term is expected to be weakly positive.
- Sidechain Torsion quantifies how realistic the sidechain angles are, i.e., how well they fit within the bounds of what we observe in the PDB. Rosetta uses Dunbrack rotamer libraries. As a probabilistic score, this term is expected to be weakly positive.
- Solvation measures how well the model buries hydrophobics and exposes hydrophilic residues, based on the Lazaridis/Karplus implicit model of water around a protein.
- Hydrogen Bond measures how many hydrogen bonds are formed and how well-formed their geometry is, based on PDB-derived statistics. This term is always negative.
How can I enable third-party cookies?
Bench requires third-party cookies to be enabled in your browser. Here’s how to enable them:
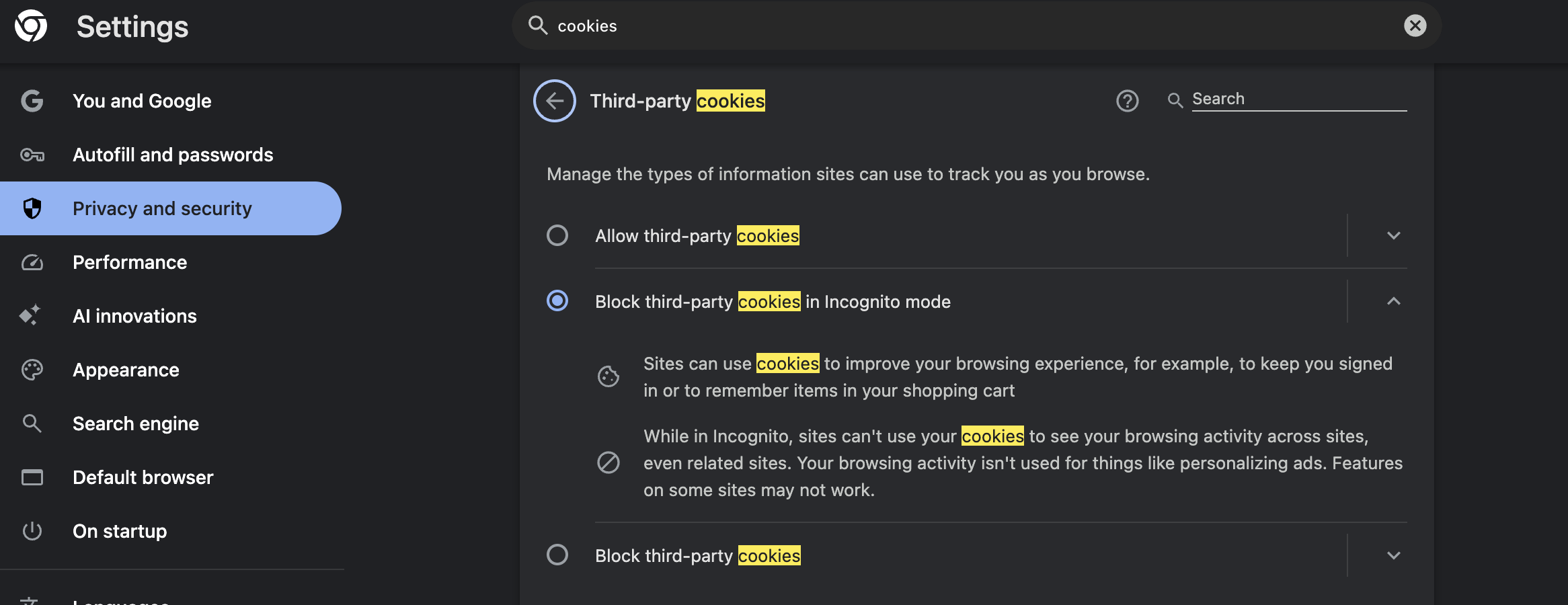
For Chrome:
- Click the three dots menu in the top right corner
- Select “Settings”
- Click “Privacy and security” in the left menu
- Click “Third-party cookies”
- Make sure “Block third-party cookies” is turned OFF
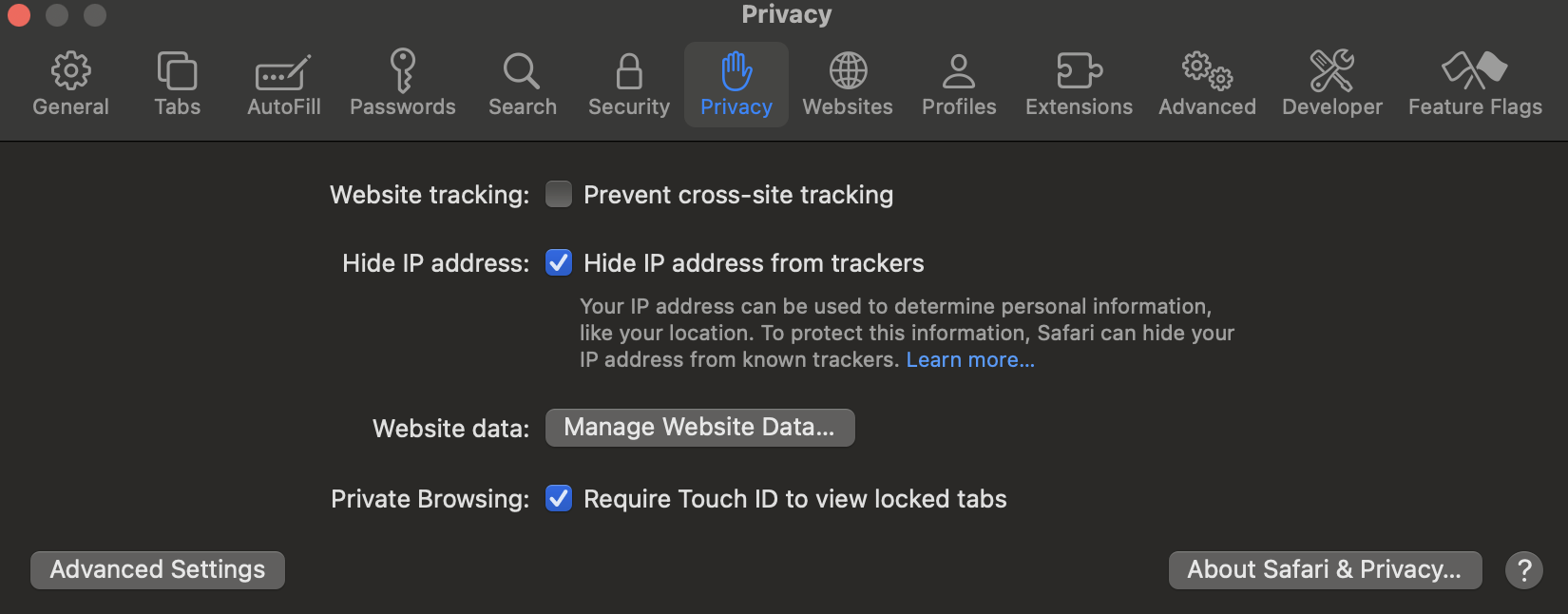
For Safari:
- Click “Safari” in the top menu bar
- Select “Settings”
- Go to the “Privacy” tab
- Uncheck “Prevent cross-site tracking”
If you’re still having issues after enabling third-party cookies, try clearing your browser cache and cookies, then restart your browser.